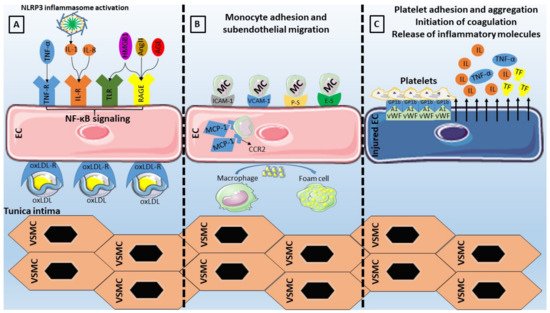
Concerning adhesion molecules specifically (
Table 1), VCAM-1 has been considered an essential component of the endothelial activation cascade as it is involved in the selective adhesiveness of monocytes and lymphocytes, as they have been found to express the counterreceptor very late antigen (VLA)-4
[96][97][96,97]. ICAM-1 is also implicated in the interaction between endothelial cells and monocytes through its ligands (lymphocyte function-associated antigen-1, macrophage-1 antigen), by enabling the adhesion and migration of leukocytes
[98]. Moving on to selectins (P-Selectin, E-Selectin, and L-Selectin), all of which can be found on the surface of endothelial cells, leukocytes, and platelets, their structure is based on an N-terminal carbohydrate recognition domain, an epidermal growth factor-like domain, a varying number of short consensus repeats that have homology to complement regulatory domains (2, 6, and 9 within L-, E-, and P-selectin respectively), a transmembrane region, and a C-terminal cytoplasmatic tail
[99]. The largest member of the family, P-Selectin, is situated on the membrane of the Weibel-Palade bodies of endothelial cells. The presence of its primary ligand (P-selectin glycoprotein ligand-1) on the surface of leukocytes indicates its role in leukocyte adhesion and rolling on endothelium, while available evidence points to an additional signaling role within the endothelium
[99]. E-Selectin, whose upregulation is based on the NF-κB binding to regulatory domains in its promoter, has the capability of retarding leukocyte rolling leading to leukocyte arrest, making it pivotal in leukocyte trafficking and inflammatory responses
[99]. The role of selectins in atherosclerotic diseases has been extensively investigated, with both molecules being present on endothelial cells of atherosclerotic plaques
[100], while E-selectin or P-selectin deficient mice displayed attenuated atherosclerosis
[101][102][101,102].Last but not least, MCP-1 has been the first reported inflammatory chemokine that is secreted from endothelial cells and monocytes, with its function consisting of leukocyte mobilization towards the subendothelium by binding on the CCR2 receptor, thus contributing to atherosclerosis
[103]. Animal studies have shown that MCP-1 or CCR2 deficiency leads to lower lipid deposition and slowing of the atherosclerotic process
[104][105][104,105], while its overexpression results in opposite effects with macrophage accumulation and atherosclerosis acceleration
[106]. Moreover, another study has reported a role of MCP-1 on endothelial cell apoptosis
[107]. Importantly, the role of adhesion molecules as markers of cardiovascular disease and incident cardiovascular risk has also been studied
[108][109][110][111][112][113][114][108,109,110,111,112,113,114].
Table 1. Clinical implications of adhesion molecules involved in the interplay between endothelial dysfunction and inflammation.
| Adhesion Molecule |
Ligand |
Role |
Clinical Significance |
| ICAM-1 |
LFA-1
Mac-1 |
Leukocyte adhesion |
ICAM-1 correlates with the incidence of CAD and carotid atherosclerosis independently of known cardiovascular risk factors [115]. |
| VCAM-1 |
VLA-4 |
Leukocyte adhesion |
Baseline VCAM-1 is increased in initially healthy middle-aged men who develop cardiovascular disease [116]. |
| E-Selectin |
ESL
PSGL-1 |
Leukocyte adhesion |
E-Selectin correlates with the incidence of CAD and carotid atherosclerosis independently of known cardiovascular risk factors [115]. |
| P-Selectin |
PSGL-1 |
Leukocyte adhesion |
Elevated P-selectin levels predict early adverse events in patients with presumed CAD [117]. |
| MCP-1 |
CCR2 |
Monocyte chemotaxis |
Association of MCP-1 with risk of incident PAD and CAD independently of traditional cardiovascular risk factors [118]. |
5.4. The Pro-Inflammatory Effect of NOX
NOX are the only family of enzymes primarily implicated in ROS generation, since other ROS regulators require an external ROS source to become involved in their formation. Therefore, their role in the initiation of the oxidative stress cascade is critical and depends upon the presence of cardiovascular risk factors. In such cases, NOX overproduction results in activation of inflammatory pathways together with eNOS uncoupling and scavenging of antioxidants
[119].
From the four described NOX expressed in EC, NOX1, NOX2, and NOX5 are implicated in vascular diseases while an antiatherogenic role is speculated for NOX4
[119]. Even though NOX exert their pro-atherogenic effects via promotion of oxidative stress and impairment of NO bioavailability, recent reports have linked NOX-derived ROS to propagate NF-κB signaling and, consequently, the release of adhesion molecules and pro-inflammatory mediators
[120]. Interestingly, exposure to oscillatory SS resulted in an upregulated expression of NOX2 via the action of sterol regulatory element binding protein 2, ultimately leading to NLRP3 inflammasome activation
[121].
5.5. Neutrophil Extracellular Traps
Neutrophil extracellular traps (NETs), consisting of nuclear chromatin, histones, and proteins of various origins, have been recently attached to the inflammatory background of atherosclerotic cardiovascular diseases
[122]. Excessive NET production may result in vascular leakage and endothelial-to-mesenchymal transition by degradation of VE-cadherin
[123], as well as in complement activation leading to endothelial injury
[124].
Several pro-atherosclerotic conditions have been characterized by NETosis including hyperglycemia, dyslipidemia, and obesity. Starting with hyperglycemia, it has been proven that NET production is highly NADPH oxidase-dependent in the setting of DM
[125], eventually resulting in endothelial injury via damage to the endothelial glycocalyx
[126]. In cases of dyslipidemia, oxLDL may act on neutrophils to enhance NET formation, with the resulting product being able to propagate endothelial dysfunction
[127]. Obesity is another condition with presumed NETosis owing to its pro-inflammatory actions and, as demonstrated in diet-induced obesity mouse models, the NET formation was assumed to be the orchestrator of endothelial dysfunction
[128]. Studies assessing the association of NETs with endothelial function in humans are scarce, however, highlighting the continuous research that ought to be performed in this field.
5.6. Shear Stress
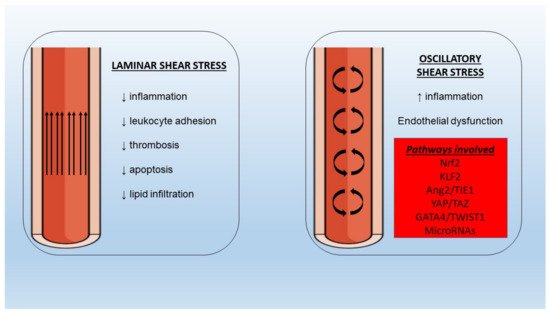
Endothelial cell regulation at the mechanical level is achieved via SS forces. In the setting of a laminar SS the beneficial, atheroprotective functions of endothelium remain intact (
Figure 2). However, in areas of bifurcations, there is a differentiation of flow pattern towards turbulence, with an altered phenotype leading to increased adhesion of monocytes, proliferation, and apoptosis. It has been shown that the application of SS leads to the activation of multiple cell membrane mechanosensors such as integrins
[129], G protein-coupled receptors
[130], and endothelial glycocalyx
[131], among others, with ensuing activation of downstream signaling pathways associated with functional gene expression. It is important to note that laminar SS application on cultured endothelial cells resulted in enhanced expression of eNOS
[132], with Akt phosphorylation and subsequent eNOS phosphorylation at Ser 1177 being important for NO production
[133].
Figure 2. Different properties of laminar and oscillatory shear stress under normal and pathologic conditions with implications for inflammation and endothelial dysfunction. Nrf2: nuclear factor erythroid 2–related factor 2, KLF2: Krüppel-like factor-2, Ang2: angiopoietin-2, TIE1: tyrosine kinase with immunoglobulin-like and EGF-like domains-1, YAP: yes-associated protein, TWIST1: twist-related protein-1.
In the presence of oscillating or low SS an increased burden of NF-κB molecules has been observed
[134], pointing to a pro-inflammatory effect on the underlying endothelium which is potentially mediated by nuclear factor erythroid 2–related factor 2 (Nrf2) and Krüppel-like factor (KLF)-2
[135][136][135,136]. Moreover, low SS could be responsible for the disturbances of angiopoietin (Ang)/tyrosine kinase with immunoglobulin-like and EGF-like domains 1 (TIE1) axis via upregulation of the pro-inflammatory Ang2 in endothelial cells
[137]. Recent evidence from omics studies of endothelial cells has proposed additional novel links between SS and genes involved in embryonic development. To begin with, the yes-associated protein (YAP)/TAZ pathway promotes JUN N-terminal kinase-mediated inflammation in conditions of low SS
[138][139][138,139]. Twist-related protein 1 (TWIST1) is another molecule involved in developmental processes that has been investigated since it was found increased in aortic areas of low SS
[140]. In a preclinical study, TWIST1 was induced in a GATA4-dependent manner after exposure to low SS with inflammation and endothelial-to-mesenchymal transition processes being also promoted, thus leading to endothelial dysfunction
[141]. Several other pathways (bone morphogenetic protein signaling, WNT signaling) have been implicated in the interaction between shear stress, inflammation, and endothelial function and merit further validation in animal studies
[142][143][142,143].
Flow alterations have important implications in microRNA regulation. MicroRNAs are small non-coding RNA molecules consisting of 18–24 nucleotides that contribute to the regulation of numerous cellular functions such as multiplication, differentiation, apoptosis, and death, especially in situations of inflammation or trauma, via RNA interference and post-translational modification in gene expression. It is, therefore, expected that deregulated expression of epigenetics will affect inflammatory pathways and endothelial cell function. Starting with microRNA-126, which is believed to be a key regulator of vascular endothelial homeostasis and inflammation
[144][145][144,145], even though early studies suggested a pro-atherogenic role, the latest evidence proposes an anti-inflammatory, endothelial-protective mechanism of action which is lost in conditions of disturbed flow
[146][147][148][146,147,148]. MicroRNA 19a, a member of the microRNA-17~92a, is overexpressed in low SS and exerts pro-inflammatory effects by targeting HMGB1
[149]. MicroRNA-92 from the same cluster is also overexpressed in disturbed flow conditions and targets KLF2, KLF4, and the suppressor of cytokine signaling 5 (SOCS5), leading to endothelial inflammation and dysfunction
[150]. Next, microRNA-663 overexpression under low SS conditions appears to induce endothelial inflammation with multiple targets involved
[151]. MicroRNA-712, solely found in murine animal models, and its human homolog microRNA-205 are upregulated in situations of disturbed flow and are involved in endothelial cell inflammation mediated by the loss of tissue inhibitor of metalloproteinase-3 (TIMP3) expression
[152]. MicroRNA-181 is also reduced under low SS conditions leading to NLR family pyrin domain containing 3 (NLRP3) inflammasome-dependent pyroptosis of endothelial cells
[153].
5.7. Endothelial Dysfunction in Chronic Inflammatory Diseases
An excess of adverse cardiovascular events in individuals with chronic autoimmune inflammatory diseases has been noted, with endothelial dysfunction potentially being the crucial link. Since these conditions are characterized by a significant inflammatory burden, the overexpression of inflammatory cytokines leads to increased oxidative stress and dyslipidemia, while the role of autoantibodies in the development of endothelial dysfunction remains to be elucidated
[154].
Starting with rheumatoid arthritis, the presence of endothelial dysfunction is frequently present, as shown in a recent meta-analysis
[155]. However, the correlations with disease duration, activity, and remission have been inconsistent
[156]. In the case of systemic lupus erythematosus, which is also accompanied by increased cardiovascular disease rates
[157], a higher prevalence of endothelial dysfunction has been observed the latest meta-analysis
[158]. The presence of endothelial dysfunction has been described in patients with psoriasis, possibly due to the reduced NO bioavailability, which is correlated to symptom severity
[159]. Importantly, the presence of impaired coronary endothelial function, assessed by positron emission tomography, in the above-mentioned inflammatory states was related to an increased rate of major adverse cardiovascular events
[160]. Lastly, impaired endothelial function has been documented in patients with inflammatory bowel disease, correlated with disease activity in the majority of studies
[161].