 +1 credit
+1 credit
Video Upload Options
This entry focuses on one of the 16 proteins composing the V-ATPase complex responsible for resorbing bone: the a3 subunit. The rationale for focusing on this biomolecule is that mutations in this one protein account for over 50% of osteopetrosis cases, highlighting its critical role in bone physiology. Despite its essential role in bone remodeling and its involvement in bone diseases, little is known about the way in which this subunit is targeted and regulated within osteoclasts. To this end, this review is broadened to include the three other mammalian paralogues (a1, a2 and a4) and the two yeast orthologs (Vph1p and Stv1p). By examining the literature on all of the paralogues/orthologs of the V-ATPase a subunit, we hope to provide insight into the molecular mechanisms and future research directions specific to a3. This review starts with an overview on bone, highlighting the role of V-ATPases in osteoclastic bone resorption. We then cover V-ATPases in other location/functions, highlighting the roles which the four mammalian a subunit paralogues might play in differential targeting and/or regulation. Author review the ways in which the energy of ATP hydrolysis is converted into proton translocation, and go in depth into the diverse role of the a subunit, not only in proton translocation but also in lipid binding, cell signaling and human diseases. Finally, the therapeutic implication of targeting a3 specifically for bone diseases and cancer is discussed, with concluding remarks on future directions.
1. Bone
2. V-ATPase Functions
3. V-ATPase Structure
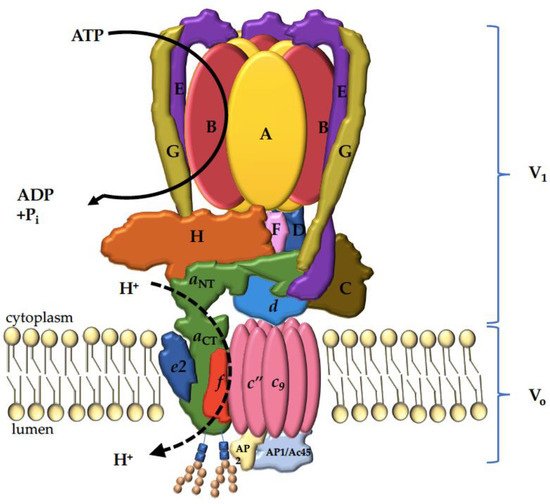
4. The V-ATPase a Subunit
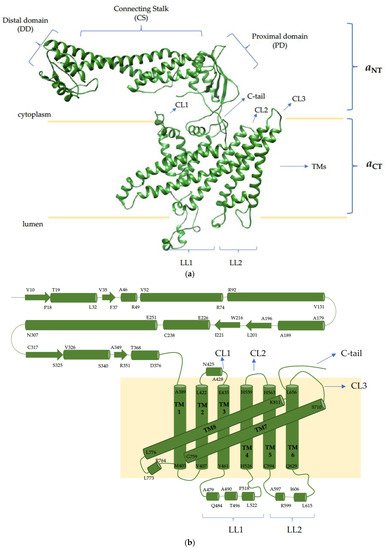
References
- Lin, X.; Patil, S.; Gao, Y.-G.; Qian, A. The Bone Extracellular Matrix in Bone Formation and Regeneration. Front. Pharmacol. 2020, 11, 757.
- Lee, D.; Kim, D.W.; Cho, J.-Y. Correction to: Role of growth factors in hematopoietic stem cell niche. Cell Biol. Toxicol. 2020, 36, 1.
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.-R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169.
- Roeder, E.; Matthews, B.G.; Kalajzic, I. Visual reporters for study of the osteoblast lineage. Bone 2016, 92, 189–195.
- Prideaux, M.; Findlay, D.M.; Atkins, G.J. Osteocytes: The master cells in bone remodelling. Curr. Opin. Pharmacol. 2016, 28, 24–30.
- Creecy, A.; Damrath, J.G.; Wallace, J.M. Control of Bone Matrix Properties by Osteocytes. Front. Endocrinol. 2021, 11, 578477.
- Chen, X.; Wang, Z.; Duan, N.; Zhu, G.; Schwarz, E.M.; Xie, C. Osteoblast-osteoclast interactions. Connect. Tissue Res. 2018, 59, 99–107.
- Jacome-Galarza, C.E.; Percin, G.I.; Muller, J.T.; Mass, E.; Lazarov, T.; Eitler, J.; Rauner, M.; Yadav, V.K.; Crozet, L.; Bohm, M.; et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nat. Cell Biol. 2019, 568, 541–545.
- Yahara, Y.; Barrientos, T.; Tang, Y.J.; Puviindran, V.; Nadesan, P.; Zhang, H.; Gibson, J.R.; Gregory, S.G.; Diao, Y.; Xiang, Y.; et al. Erythromyeloid progenitors give rise to a population of osteoclasts that contribute to bone homeostasis and repair. Nat. Cell Biol. 2020, 22, 49–59.
- Søe, K.; Delaisse, J.-M.; Borggaard, X.G. Osteoclast formation at the bone marrow/bone surface interface: Importance of structural elements, matrix, and intercellular communication. Semin. Cell Dev. Biol. 2020, 112, 8–15.
- Sims, N.A.; Martin, T.J. Osteoclasts Provide Coupling Signals to Osteoblast Lineage Cells Through Multiple Mechanisms. Annu. Rev. Physiol. 2020, 82, 507–529.
- Takito, J.; Nakamura, M. Heterogeneity and Actin Cytoskeleton in Osteoclast and Macrophage Multinucleation. Int. J. Mol. Sci. 2020, 21, 6629.
- Gambari, L.; Grassi, F.; Roseti, L.; Grigolo, B.; Desando, G. Learning from Monocyte-Macrophage Fusion and Multinucleation: Potential Therapeutic Targets for Osteoporosis and Rheumatoid Arthritis. Int. J. Mol. Sci. 2020, 21, 6001.
- Takito, J.; Inoue, S.; Nakamura, M. The Sealing Zone in Osteoclasts: A Self-Organized Structure on the Bone. Int. J. Mol. Sci. 2018, 19, 984.
- Mulari, M.T.K.; Zhao, H.; Lakkakorpi, P.T.; Väänänen, H.K. Osteoclast Ruffled Border Has Distinct Subdomains for Secretion and Degraded Matrix Uptake. Traffic 2003, 4, 113–125.
- Toyomura, T.; Murata, Y.; Yamamoto, A.; Oka, T.; Sun-Wada, G.H.; Wada, Y.; Futai, M. From lysosomes to the plasma membrane: Localization of vacuolar-type H+ -ATPase with the a3 isoform during osteoclast differentiation. J. Biol. Chem. 2003, 278, 22023–22030.
- Ng, P.Y.; Ribet, A.B.P.; Pavlos, N.J. Membrane trafficking in osteoclasts and implications for osteoporosis. Biochem. Soc. Trans. 2019, 47, 639–650.
- Kim, J.H.; Kim, N. Bone Cell Communication Factors Provide a New Therapeutic Strategy for Osteoporosis. Chonnam. Med. J. 2020, 56, 94–98.
- Wang, H.; Yang, G.; Xiao, Y.; Luo, G.; Li, G.; Li, Z. Friend or Foe? Essential Roles of Osteoclast in Maintaining Skeletal Health. BioMed Res. Int. 2020, 2020, 1–10.
- Corbacho, I.; Teixidó, F.; Olivero, I.; Hernández, L.M. Dependence of Saccharomyces cerevisiae Golgi functions on V-ATPase activity. FEMS Yeast Res. 2012, 12, 341–350.
- Nelson, N. Structure and function of V-ATPases in endocytic and secretory organelles. J. Exp. Biol. 1992, 172, 149–153.
- Perzov, N.; Padler-Karavani, V.; Nelson, H.; Nelson, N. Characterization of yeast V-ATPase mutants lacking Vph1p or Stv1p and the effect on endocytosis. J. Exp. Biol. 2002, 205, 1209–1219.
- Márquez-Sterling, N.; Herman, I.M.; Pesacreta, T.; Arai, H.; Terres, G.; Forgac, M. Immunolocalization of the vacuolar-type (H+)-ATPase from clathrin-coated vesicles. Eur. J. Cell Biol. 1991, 56, 19–33.
- Kissing, S.; Hermsen, C.; Repnik, U.; Nesset, C.K.; von Bargen, K.; Griffiths, G.; Ichihara, A.; Lee, B.S.; Schwake, M.; De Brabander, J.; et al. Vacuolar ATPase in Phagosome-Lysosome Fusion. J. Biol. Chem. 2015, 290, 14166–14180.
- Sun-Wada, G.-H.; Wada, Y. Role of vacuolar-type proton ATPase in signal transduction. Biochim. et Biophys. Acta (BBA) Bioenerg. 2015, 1847, 1166–1172.
- Stransky, L.; Cotter, K.; Forgac, M. The Function of V-ATPases in Cancer. Physiol. Rev. 2016, 96, 1071–1091.
- Kibak, H.; Taiz, L.; Starke, T.; Bernasconi, P.; Gogarten, J.P. Evolution of structure and function of V-ATPases. J. Bioenerg. Biomembr. 1992, 24, 415–424.
- Sun-Wada, G.-H.; Wada, Y.; Futai, M. Vacuolar H+ pumping ATPases in luminal acidic organelles and extracellular compartments: Common rotational mechanism and diverse physiological roles. J. Bioenerg. Biomembr. 2003, 35, 347–358.
- Futai, M.; Oka, T.; Sun-Wada, G.; Moriyama, Y.; Kanazawa, H.; Wada, Y. Luminal acidification of diverse organelles by V-ATPase in animal cells. J. Exp. Biol. 2000, 203, 107–116.
- Esmail, S.; Kartner, N.; Yao, Y.; Kim, J.W.; Reithmeier, R.A.; Manolson, M.F. Molecular mechanisms of cutis laxa– and distal renal tubular acidosis–causing mutations in V-ATPase a subunits, ATP6V0A2 and ATP6V0A4. J. Biol. Chem. 2018, 293, 2787–2800.
- Mindell, J.A. Lysosomal Acidification Mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86.
- Futai, M.; Sun-Wada, G.H.; Wada, Y.; Matsumoto, N.; Nakanishi-Matsui, M. Vacuolar-type ATPase: A proton pump to lysosomal trafficking. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 17.
- Lu, X.; Meima, M.E.; Nelson, J.K.; Sorrentino, V.; Loregger, A.; Scheij, S.; Dekkers, D.H.; Mulder, M.T.; Demmers, J.A.; M-Dallinga-Thie, G.; et al. Identification of the (Pro)renin Receptor as a Novel Regulator of Low-Density Lipoprotein MetabolismNovelty and Significance. Circ. Res. 2016, 118, 222–229.
- Yeganeh, B.; Ghavami, S.; Kroeker, A.L.; Mahood, T.H.; Stelmack, G.L.; Klonisch, T.; Coombs, K.; Halayko, A.J. Suppression of influenza A virus replication in human lung epithelial cells by noncytotoxic concentrations bafilomycin A1. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L270–L286.
- Müller, K.H.; E Kainov, D.; El Bakkouri, K.; Saelens, X.; De Brabander, J.K.; Kittel, C.; Samm, E.; Muller, C.P. The proton translocation domain of cellular vacuolar ATPase provides a target for the treatment of influenza A virus infections. Br. J. Pharmacol. 2011, 164, 344–357.
- Gary-Bobo, M.; Nirdé, P.; Jeanjean, A.; Morère, A.; Garcia, M. Mannose 6-Phosphate Receptor Targeting and its Applications in Human Diseases. Curr. Med. Chem. 2007, 14, 2945–2953.
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813.
- Efeyan, A.; Zoncu, R.; Sabatini, D.M. Amino acids and mTORC1: From lysosomes to disease. Trends Mol. Med. 2012, 18, 524–533.
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 Senses Lysosomal Amino Acids Through an Inside-Out Mechanism That Requires the Vacuolar H+-ATPase. Science 2011, 334, 678–683.
- Stransky, L.A.; Forgac, M. Amino Acid Availability Modulates Vacuolar H+-ATPase Assembly. J. Biol. Chem. 2015, 290, 27360–27369.
- Rama, S.; Boumedine-Guignon, N.; Sangiardi, M.; Youssouf, F.; Maulet, Y.; Lévêque, C.; Belghazi, M.; Seagar, M.; Debanne, D.; El Far, O. Chromophore-Assisted Light Inactivation of the V-ATPase V0c Subunit Inhibits Neurotransmitter Release Downstream of Synaptic Vesicle Acidification. Mol. Neurobiol. 2018, 56, 3591–3602.
- Morel, N.; Poëa-Guyon, S. The membrane domain of vacuolar H+ATPase: A crucial player in neurotransmitter exocytotic release. Cell. Mol. Life Sci. 2015, 72, 2561–2573.
- Morel, N. Neurotransmitter release: The dark side of the vacuolar-H+ATPase. Biol. Cell 2003, 95, 453–457.
- Marshansky, V.R.; Grüber, G. Eukaryotic V-ATPase: Novel structural findings and functional insights. Biochim. Biophys. Acta 2014, 1837, 23.
- Sun-Wada, G.H.; Toyomura, T.; Murata, Y.; Yamamoto, A.; Futai, M.; Wada, Y. The a3 isoform of V-ATPase regulates insulin secretion from pancreatic beta-cells. J. Cell. Sci. 2006, 119, 4531–4540.
- Sun-Wada, G.-H.; Tabata, H.; Kawamura, N. Selective Assembly of V-ATPase Subunit Isoforms in Mouse Kidney. J. Bioenerg. Biomembr. 2005, 37, 415–418.
- Brown, D.; Sabolic, I.; Gluck, S. Polarized targeting of V-ATPase in kidney epithelial cells. J. Exp. Biol. 1992, 172, 231–243.
- Hermo, L.; I Adamali, H.; Andonian, S. Immunolocalization of CA II and H+ V-ATPase in epithelial cells of the mouse and rat epididymis. J. Androl. 2000, 21, 376–391.
- Pietrement, C.; Sun-Wada, G.-H.; Da Silva, N.; McKee, M.; Marshansky, V.; Brown, D.; Futai, M.; Breton, S. Distinct Expression Patterns of Different Subunit Isoforms of the V-ATPase in the Rat Epididymis1. Biol. Reprod. 2006, 74, 185–194.
- Qin, A.; Cheng, T.; Pavlos, N.; Lin, Z.; Dai, K.; Zheng, M. V-ATPases in osteoclasts: Structure, function and potential inhibitors of bone resorption. Int. J. Biochem. Cell Biol. 2012, 44, 1422–1435.
- Kartner, N.; Manolson, M. V-ATPase subunit interactions: The long road to therapeutic targeting. Curr. Protein Pept. Sci. 2012, 13, 164–179.
- Hinton, A.; Bond, S.; Forgac, M. V-ATPase functions in normal and disease processes. Pflügers Arch. Eur. J. Physiol. 2009, 457, 589–598.
- Frattini, A.; Orchard, P.J.; Sobacchi, C.; Giliani, S.; Abinun, M.; Mattsson, J.P.; Keeling, D.J.; Andersson, A.-K.; Wallbrandt, P.; Zecca, L.; et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat. Genet. 2000, 25, 343–346.
- Nakhoul, N.L.; Hamm, L.L. Vacuolar H(+)-ATPase in the kidney. J. Nephrol. 2002, 15, 22–31.
- Kartner, N.; Yao, Y.; Li, K.; Crasto, G.J.; Datti, A.; Manolson, M.F. Inhibition of Osteoclast Bone Resorption by Disrupting Vacuolar H+-ATPase a3-B2 Subunit Interaction. J. Biol. Chem. 2010, 285, 37476–37490.
- Blair, H.C.; Teitelbaum, S.L.; Ghiselli, R.; Gluck, S. Osteoclastic bone resorption by a polarized vacuolar proton pump. Science 1989, 245, 855–857.
- Collins, M.P.; Forgac, M. Regulation of V-ATPase Assembly in Nutrient Sensing and Function of V-ATPases in Breast Cancer Metastasis. Front. Physiol. 2018, 9, 902.
- Michel, V.; Munoz, Y.L.; Trujillo, K.; Bisoffi, M.; Parra, K.J. Inhibitors of vacuolar ATPase proton pumps inhibit human prostate cancer cell invasion and prostate-specific antigen expression and secretion. Int. J. Cancer 2013, 132, E1–E10.
- Jung, Y.S.; Jun, S.; Kim, M.J.; Lee, S.H.; Suh, H.N.; Lien, E.M.; Jung, H.Y.; Lee, S.; Zhang, J.; Yang, J.I.; et al. TMEM9 promotes intestinal tumorigenesis through vacuolar-ATPase-activated Wnt/beta-catenin signalling. Nat. Cell Biol. 2018, 20, 1421–1433.
- Wang, D.; Epstein, D.; Khalaf, O.; Srinivasan, S.; Williamson, W.R.; Fayyazuddin, A.; Quiocho, F.A.; Hiesinger, P.R. Ca2+-Calmodulin regulates SNARE assembly and spontaneous neurotransmitter release via v-ATPase subunit V0a1. J. Cell Biol. 2014, 205, 21–31.
- Portela, M.; Yang, L.; Paul, S.; Li, X.; Veraksa, A.; Parsons, L.M.; Richardson, H.E. Lgl reduces endosomal vesicle acidification and Notch signaling by promoting the interaction between Vap33 and the V-ATPase complex. Sci. Signal. 2018, 11, eaar1976.
- Abbas, Y.M.; Wu, D.; Bueler, S.A.; Robinson, C.V.; Rubinstein, J.L. Structure of V-ATPase from the mammalian brain. Science 2020, 367, 1240–1246.
- Jansen, E.J.; Scheenen, W.J.; Hafmans, T.G.; Martens, G.J. Accessory subunit Ac45 controls the V-ATPase in the regulated secretory pathway. Biochim. Biophys. Acta (BBA) Bioenerg. 2008, 1783, 2301–2310.
- Mazhab-Jafari, M.T.; Rohou, A.; Schmidt, C.; Bueler, S.A.; Benlekbir, S.; Robinson, C.; Rubinstein, J.L. Atomic model for the membrane-embedded VO motor of a eukaryotic V-ATPase. Nat. Cell Biol. 2016, 539, 118–122.
- Kawasaki-Nishi, S.; Nishi, T.; Forgac, M. Proton translocation driven by ATP hydrolysis in V-ATPases. FEBS Lett. 2003, 545, 76–85.
- Esmail, S.; Kartner, N.; Yao, Y.; Kim, J.W.; Reithmeier, R.A.F.; Manolson, M.F. N-linked glycosylation of a subunit isoforms is critical for vertebrate vacuolar H(+) -ATPase (V-ATPase) biosynthesis. J. Cell Biochem. 2018, 119, 861–875.
- Flannery, A.R.; Graham, L.A.; Stevens, T.H. Topological Characterization of the c, c′, and c″ Subunits of the Vacuolar ATPase from the Yeast Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 39856–39862.
- Wang, Y.; Cipriano, D.J.; Forgac, M. Arrangement of Subunits in the Proteolipid Ring of the V-ATPase. J. Biol. Chem. 2007, 282, 34058–34065.
- Kawasaki-Nishi, S.; Nishi, T.; Forgac, M. Arg-735 of the 100-kDa subunit a of the yeast V-ATPase is essential for proton translocation. Proc. Natl. Acad. Sci. USA 2001, 98, 12397–12402.
- Oot, R.A.; Kane, P.M.; Berry, E.A.; Wilkens, S. Crystal structure of yeast V1-ATPase in the autoinhibited state. EMBO J. 2016, 35, 1694–1706.
- Smardon, A.M.; Tarsio, M.; Kane, P.M. The RAVE Complex Is Essential for Stable Assembly of the Yeast V-ATPase. J. Biol. Chem. 2002, 277, 13831–13839.
- Kane, P.M. Targeting Reversible Disassembly as a Mechanism of Controlling V-ATPase Activity. Curr. Protein Pept. Sci. 2012, 13, 117–123.
- Sharma, S.; Oot, R.A.; Wilkens, S. MgATP hydrolysis destabilizes the interaction between subunit H and yeast V1-ATPase, highlighting H’s role in V-ATPase regulation by reversible disassembly. J. Biol. Chem. 2018, 293, 10718–10730.
- Lu, M.; Holliday, L.S.; Zhang, L.; Dunn, W.A., Jr.; Gluck, S.L. Interaction between aldolase and vacuolar H+-ATPase: Evidence for direct coupling of glycolysis to the ATP-hydrolyzing proton pump. J. Biol. Chem. 2001, 276, 30407–30413.
- Lu, M.; Sautin, Y.; Holliday, L.S.; Gluck, S.L. The Glycolytic Enzyme Aldolase Mediates Assembly, Expression, and Activity of Vacuolar H+-ATPase. J. Biol. Chem. 2004, 279, 8732–8739.
- Li, M.; Zhang, C.-S.; Zong, Y.; Feng, J.-W.; Ma, T.; Hu, M.; Lin, Z.; Li, X.; Xie, C.; Wu, Y.; et al. Transient Receptor Potential V Channels Are Essential for Glucose Sensing by Aldolase and AMPK. Cell Metab. 2019, 30, 508–524.
- Banerjee, S.; Kane, P.M. Regulation of V-ATPase Activity and Organelle pH by Phosphatidylinositol Phosphate Lipids. Front. Cell Dev. Biol. 2020, 8, 510.
- Fogarty, F.M.; O’Keeffe, J.; Zhadanov, A.; Papkovsky, D.; Ayllón, V.; O’Connor, R. HRG-1 enhances cancer cell invasive potential and couples glucose metabolism to cytosolic/extracellular pH gradient regulation by the vacuolar-H+ ATPase. Oncogene 2013, 33, 4653–4663.
- Marshansky, V.; Futai, M. The V-type H+-ATPase in vesicular trafficking: Targeting, regulation and function. Curr. Opin. Cell Biol. 2008, 20, 415–426.
- Qi, J.; Wang, Y.; Forgac, M. The vacuolar (H+)-ATPase: subunit arrangement and in vivo regulation. J. Bioenerg. Biomembr. 2007, 39, 423–426.
- Rahman, S.; Yamato, I.; Murata, T. Function and Regulation of Mammalian V-ATPase Isoforms. In Regulation of Ca2+-ATPases, V-ATPases and F-ATPases; Springer Science and Business Media LLC: Cham, Switzerland, 2016; Volume 14, pp. 283–299.
- Lafourcade, C.; Sobo, K.; Kieffer-Jaquinod, S.; Garin, J.; Van Der Goot, F.G. Regulation of the V-ATPase along the Endocytic Pathway Occurs through Reversible Subunit Association and Membrane Localization. PLoS ONE 2008, 3, e2758.
- Manolson, M.F.; Wu, B.; Proteau, D.; Taillon, B.E.; Roberts, B.T.; Hoyt, M.A.; Jones, E.W. STV1 gene encodes functional homologue of 95-kDa yeast vacuolar H(+)-ATPase subunit Vph1p. J. Biol. Chem. 1994, 269, 14064–14074.
- Srinivasan, S.; Vyas, N.K.; Baker, M.L.; Quiocho, F.A. Erratum to “Crystal Structure of the Cytoplasmic N-Terminal Domain of Subunit I, a Homolog of Subunit a, of V-ATPase” [J. Mol. Biol. 412/1 (2011) 14–21]. J. Mol. Biol. 2011, 413, 523.
- Kawasaki-Nishi, S.; Nishi, T.; Forgac, M.; Garofano, A.; Zwicker, K.; Kerscher, S.; Okun, P.; Brandt, U. Interacting Helical Surfaces of the Transmembrane Segments of Subunits a and c′ of the Yeast V-ATPase Defined by Disulfide-mediated Cross-linking. J. Biol. Chem. 2003, 278, 41908–41913.
- Ochotny, N.; Flenniken, A.M.; Owen, C.; Voronov, I.; A Zirngibl, R.; Osborne, L.R.; E Henderson, J.; Adamson, S.L.; Rossant, J.; Manolson, M.; et al. The V-ATPase a3 subunit mutation R740S is dominant negative and results in osteopetrosis in mice. J. Bone Miner. Res. 2011, 26, 1484–1493.
- Kawasaki-Nishi, S.; Bowers, K.; Nishi, T.; Forgac, M.; Stevens, T.H. The Amino-terminal Domain of the Vacuolar Proton-translocating ATPase a Subunit Controls Targeting and in Vivo Dissociation, and the Carboxyl-terminal Domain Affects Coupling of Proton Transport and ATP Hydrolysis. J. Biol. Chem. 2001, 276, 47411–47420.
- Finnigan, G.C.; Cronan, G.E.; Park, H.J.; Srinivasan, S.; Quiocho, F.A.; Stevens, T.H. Sorting of the yeast vacuolar-type, proton-translocating ATPase enzyme complex (V-ATPase): Identification of a necessary and sufficient Golgi/endosomal retention signal in Stv1p. J. Biol. Chem. 2012, 287, 19487–194500.
- Zhang, W.; Wang, D.; Volk, E.; Bellen, H.J.; Hiesinger, P.R.; Quiocho, F.A. V-ATPase V0 sector subunit a1 in neurons is a target of calmodulin. J. Biol. Chem. 2008, 283, 294–300.
- Saw, N.M.N.; Kang, S.-Y.A.; Parsaud, L.; Han, G.A.; Jiang, T.; Grzegorczyk, K.; Surkont, M.; Sun-Wada, G.-H.; Wada, Y.; Li, L.; et al. Vacuolar H+-ATPase subunits Voa1 and Voa2 cooperatively regulate secretory vesicle acidification, transmitter uptake, and storage. Mol. Biol. Cell 2011, 22, 3394–3409.
- Matsumoto, N.; Sekiya, M.; Tohyama, K.; Ishiyama-Matsuura, E.; Sun-Wada, G.-H.; Wada, Y.; Futai, M.; Nakanishi-Matsui, M. Essential Role of the a3 Isoform of V-ATPase in Secretory Lysosome Trafficking via Rab7 Recruitment. Sci. Rep. 2018, 8, 1–18.
- Matsumoto, N.; Daido, S.; Sun-Wada, G.-H.; Wada, Y.; Futai, M.; Nakanishi-Matsui, M. Diversity of proton pumps in osteoclasts: V-ATPase with a3 and d2 isoforms is a major form in osteoclasts. Biochim. et Biophys. Acta (BBA) Bioenerg. 2014, 1837, 744–749.
- Oka, T.; Murata, Y.; Namba, M.; Yoshimizu, T.; Toyomura, T.; Yamamoto, A.; Sun-Wada, G.-H.; Hamasaki, N.; Wada, Y.; Futai, M. a4, a Unique Kidney-specific Isoform of Mouse Vacuolar H+-ATPase Subunit a. J. Biol. Chem. 2001, 276, 40050–40054.
- McGuire, C.M.; Collins, M.P.; Sun-Wada, G.; Wada, Y.; Forgac, M. Isoform-specific gene disruptions reveal a role for the V-ATPase subunit a4 isoform in the invasiveness of 4T1-12B breast cancer cells. J. Biol. Chem. 2019, 294, 11248–11258.
- Toyomura, T.; Oka, T.; Yamaguchi, C.; Wada, Y.; Futai, M. Three subunit a isoforms of mouse vacuolar H(+)-ATPase. Preferential expression of the a3 isoform during osteoclast differentiation. J. Biol. Chem. 2000, 275, 8760–8765.
- Kornak, U.; Schulz, A.; Friedrich, W.; Uhlhaas, S.; Kremens, B.; Voit, T.; Hasan, C.; Bode, U.; Jentsch, T.J.; Kubisch, C. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum. Mol. Genet. 2000, 9, 2059–2063.
- Zirngibl, R.A.; Wang, A.; Yao, Y.; Manolson, M.; Krueger, J.; Dupuis, L.; Mendoza-Londono, R.; Voronov, I. Novel c.G630A TCIRG1 mutation causes aberrant splicing resulting in an unusually mild form of autosomal recessive osteopetrosis. J. Cell. Biochem. 2019, 120, 17180–17193.
- Duan, X.; Yang, S.; Zhang, L.; Yang, T. V-ATPases and osteoclasts: Ambiguous future of V-ATPases inhibitors in osteoporosis. Theranostics 2018, 8, 5379–5399.
- Kartner, N.; Manolson, M.F. The Vacuolar Proton ATPase (V-ATPase): Regulation and Therapeutic Targeting. In Regulation of Ca2+-ATPases, V-ATPases and F-ATPases, 1st ed.; Chakraborti, S., Dhalla, N.S., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 14, pp. 407–437.


