This rentryview focuses on one of the 16 proteins composing the V-ATPase complex responsible for resorbing bone: the a3 subunit. The rationale for focusing on this biomolecule is that mutations in this one protein account for over 50% of osteopetrosis cases, highlighting its critical role in bone physiology. Despite its essential role in bone remodeling and its involvement in bone diseases, little is known about the way in which this subunit is targeted and regulated within osteoclasts. To this end, this review is broadened to include the three other mammalian paralogues (a1, a2 and a4) and the two yeast orthologs (Vph1p and Stv1p). By examining the literature on all of the paralogues/orthologs of the V-ATPase a subunit, we hope to provide insight into the molecular mechanisms and future research directions specific to a3. This review starts with an overview on bone, highlighting the role of V-ATPases in osteoclastic bone resorption. We then cover V-ATPases in other location/functions, highlighting the roles which the four mammalian a subunit paralogues might play in differential targeting and/or regulation. AuthorWe review the ways in which the energy of ATP hydrolysis is converted into proton translocation, and go in depth into the diverse role of the a subunit, not only in proton translocation but also in lipid binding, cell signaling and human diseases. Finally, the therapeutic implication of targeting a3 specifically for bone diseases and cancer is discussed, with concluding remarks on future directions.
- V-ATPase
- osteoclasts
- bone
- osteoporosis
- osteopetrosis
- anti-resorptive therapeutics
- signalosome
- TCIRG1
- V-type proton ATPase 116 kDa subunit a3
- OC-116 kDa
- ATP6V0A3
- ATP6V1C
1. Bone
2. V-ATPase Functions
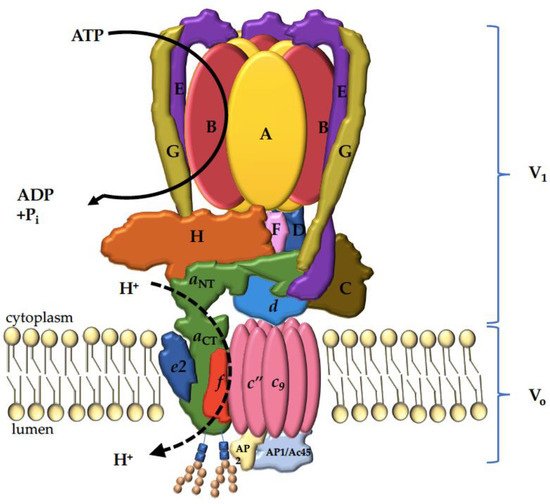
3. V-ATPase Structure
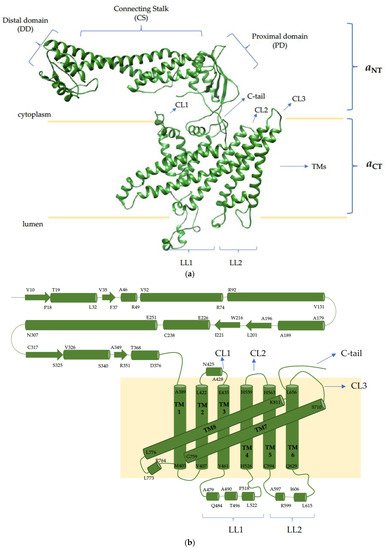
4. The V-ATPase a Subunit