Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nicolas Bonadies | + 845 word(s) | 845 | 2021-07-09 08:21:41 | | | |
| 2 | Lily Guo | Meta information modification | 845 | 2021-07-19 11:25:19 | | | | |
| 3 | Conner Chen | Meta information modification | 845 | 2021-09-22 04:15:01 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bonadies, N. MDS. Encyclopedia. Available online: https://encyclopedia.pub/entry/12170 (accessed on 07 February 2026).
Bonadies N. MDS. Encyclopedia. Available at: https://encyclopedia.pub/entry/12170. Accessed February 07, 2026.
Bonadies, Nicolas. "MDS" Encyclopedia, https://encyclopedia.pub/entry/12170 (accessed February 07, 2026).
Bonadies, N. (2021, July 19). MDS. In Encyclopedia. https://encyclopedia.pub/entry/12170
Bonadies, Nicolas. "MDS." Encyclopedia. Web. 19 July, 2021.
Copy Citation
Myelodysplastic syndromes (MDS) represent a heterogeneous group of clonal disorders caused by sequential accumulation of somatic driver mutations in hematopoietic stem and progenitor cells (HSPCs). MDS is characterized by ineffective hematopoiesis with cytopenia, dysplasia, inflammation, and a variable risk of transformation into secondary acute myeloid leukemia.
myelodysplastic syndromes
postgenomic era
precision medicine
targeted therapies
future perspectives
1. Epidemiology
MDS is a heterogeneous group of clonal conditions arising from somatic mutations in hematopoietic stem and progenitor cells (HSPCs), mainly affecting elderly individuals [1]. Ineffective hematopoiesis in MDS is characterized by a vicious circle of maturation defects (dysplasia), inflammation in the bone marrow (BM) microenvironment and cytopenia in peripheral blood (PB), which is accompanied by variable risk of progressing towards secondary Acute Myeloid Leukemia (sAML) [2]. The median age at presentation is above 70 years, with an age-standardized incidence-rate of 3–5 cases per 100,000 patient-years and a prevalence of 20 patients per 100,000 individuals [3]. The age-specific incidence-rate increases progressively with age, with 50 cases per 100,000 patient-years in individuals 75 years [3][4]. Males are predominantly affected, with the exception of MDS with isolated del(5q), which is more frequent in females. Therapy-related MDS is estimated to represent 10% of all MDS cases, though precise incidence rates cannot be determined from current epidemiological data [5][6]. Although generally a disease of the elderly, MDS can occur at any age. The presence of genetic predisposition syndromes should be thoroughly investigated in childhood or younger adults (40 years). In such cases, multiple organs can be affected, and these individuals carry a risk for increased toxicity to chemotherapy and development of other cancers [7][8].
2. General Aspects of MDS Patient Management
The heterogeneity of MDS and the multimorbidity represent major challenges. The disease course may vary from chronic asymptomatic or minimal symptomatic cytopenia to rapid progression towards sAML. Therefore, correct diagnosis, disease- and patient-based risk stratification are essential for an appropriate treatment plan. Experienced physicians, acting within interdisciplinary diagnostic and therapy review boards, should preferentially be involved. Lower-risk MDS patients have a median survival of 3 to 8 years and mostly succumb to non-leukemic causes of death. These include mainly cardiovascular events, infections and other relevant comorbidities, being aggravated by cytopenia and inflammation. Thus, treatment in lower-risk MDS should improve symptomatic cytopenia and optimize comorbidities aiming to improve quality of life (QoL) and delay progression [9][10]. Higher-risk MDS patients have a median survival of 1 to 3 years and die predominantly of complications related to sAML progression. The treatment aim in these patients is the reduction of progression and improvement of overall survival (OS) with minimal treatment-related toxicities [11][12].
3. Diagnostic Approach and Risk-Stratification
In patients with suspected MDS, previous exposure to genotoxic agents (e.g., cytotoxic chemotherapy, radiation) should be evaluated, which indicates the presence of a therapy related myeloid neoplasm (t-MN). Younger MDS patients (40 years) should be thoroughly screened for germline predispositions, which may be indicated by a family history of malignancies as well as immune or organ dysfunctions in first- and second-degree relatives. The European Leukemia Net recommendation recognizes diagnostic procedures as “mandatory” (evaluation of PB smears and BM aspirate/biopsy with cytogenetic analysis), “recommended” (fluorescence in situ hybridization (FISH) and flow cytometry), and “suggested” in specific circumstances (single-nucleotide-polymorphism (SNP), molecular diagnostics) [13]. Process-based indicators as measurable elements for quality of care are of increasing interest to enable assessment and comparison of the impact of different health care environments on relevant MDS outcomes [14].
4. Therapeutic Approach
Experienced physician and interdisciplinary boards should be involved in the assessment of MDS patient with symptomatic cytopenia or unexplained inflammatory conditions. This is important due to disease complexity, interfering comorbidities, and timely selection of higher-risk MDS patients being eligible for allo-HCT. Fluctuating cytopenias may be the initial manifestation of clonal hematopoiesis. However, patients with uncharacterized systemic autoinflammatory manifestations may also present with SLADMs at high VAFs, as primary manifestation of clonal hematopoiesis or even MDS [15]. New therapeutic options and clinical trials are urgently needed, especially in elderly patients with refractory conditions [16]. Therefore, symptomatic patients should be referred to experienced MDS centers, included in prospective registries/biobanks, and offered participation to clinical trials, whenever possible. The therapeutic approaches for lower- and higher-risk MDS are summarized in Figure 1A,B, respectively, and an overview on the treatment landscape can be found in Figure 2.
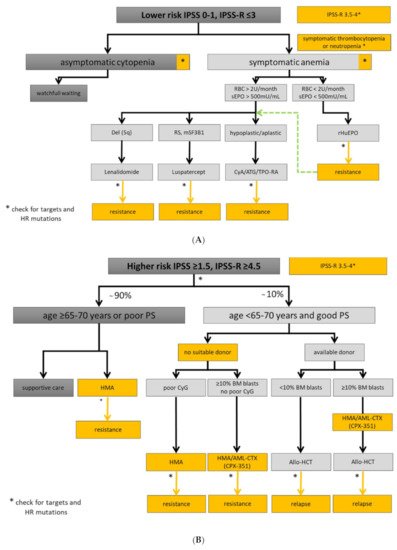
Figure 1. Treatment algorithm for lower- and higher-risk MDS. Yellow boxes highlight areas with an unmet clinical need in in lower (A) and higher-risk MDS (B). High-risk (HR) mutations comprise: ≥3 SLADMs or single mutations in TP53, RUNX1, ASXL1, ETV6, EZH2, SRSF2, U2AF1, RAS-pathway and JAK2 with VAF ≥2% [17][18][19]. Allo-HCT: allogeneic hematopoietic stem cell transplantation; AML-CTX: AML-based chemotherapy; ATG: antithymocyte globulin; BM: bone marrow; CSA: Cyclosporine A; CyG: cytogenetics; ESA: erythropoietin stimulating agent; HMA: hypomethylating agent; HR: high-risk mutations; mSF3B1: mutated SF3B1; PB: peripheral blood; PS: performance status; RBC: red blood cell concentrate; RS: ring sideroblasts; sEpo: serum erythropoietin; TPO-RA: thrombopoietin receptor agonist. Adapted from [20].
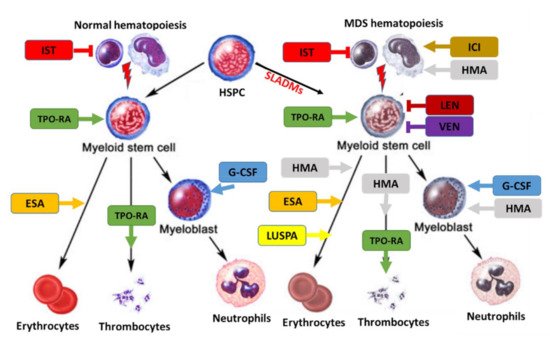
Figure 2. Treatment landscape in MDS. Treatment can affect normal (left) and clonal hematopoiesis (right). SLADMs: somatic leukemia-associated driver mutations; ESA, erythropoietin stimulating agent; ICI, immune checkpoint inhibitors; G-CSF, granulocyte colony stimulating factor; HMA, hypomethylating agents; IST, immune suppressive treatment (CyA/ATG); LEN, lenalidomide; LUSPA, luspatercept; TPO-RA: thrombopoietin receptor agonists; VEN, venetoclax. Adapted from [20].
References
- Shastri, A.; Will, B.; Steidl, U.; Verma, A. Stem and progenitor cell alterations in myelodysplastic syndromes. Blood 2017, 129, 1586–1594.
- da Silva-Coelho, P.; Kroeze, L.I.; Yoshida, K.; Koorenhof-Scheele, T.N.; Knops, R.; Van De Locht, L.T.; De Graaf, A.O.; Massop, M.; Sandmann, S.; Dugas, M.; et al. Clonal evolution in myelodysplastic syndromes. Nat. Commun. 2017, 8, 15099.
- Zeidan, A.M.; Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X. Epidemiology of myelodysplastic syndromes: Why characterizing the beast is a prerequisite to taming it. Blood Rev. 2019, 34, 1–15.
- Bonadies, N.; Feller, A.; Rovo, A.; Ruefer, A.; Blum, S.; Gerber, B.; Stuessi, G.; Benz, R.; Cantoni, N.; Holbro, A.; et al. Trends of classification, incidence, mortality, and survival of MDS patients in Switzerland between 2001 and 2012. Cancer Epidemiol. 2017, 46, 85–92.
- Candelaria, M.; Dueñas-Gonzalez, A. Therapy-related myelodysplastic syndrome. Expert Opin. Drug Saf. 2015, 14, 655–665.
- Abou Zahr, A.; Kavi, A.M.; Mukherjee, S.; Zeidan, A.M. Therapy-related myelodysplastic syndromes, or are they? Blood Rev. 2017, 31, 119–128.
- Locatelli, F.; Strahm, B. How I treat myelodysplastic syndromes of childhood. Blood 2018, 131, 1406–1414.
- Stauder, R.; Yu, G.; Koinig, K.A.; Bagguley, T.; Fenaux, P.; Symeonidis, A.; Sanz, G.; Cermak, J.; Mittelman, M.; Hellström-Lindberg, E.; et al. Health-related quality of life in lower-risk MDS patients compared with age- and sex-matched reference populations: A European LeukemiaNet study. Leukemia 2018, 32, 1380–1392.
- Santini, V. Treatment of low-risk myelodysplastic syndromes. Hematology 2016, 2016, 462–469.
- Fenaux, P.; Ades, L. How we treat lower-risk myelodysplastic syndromes. Blood 2013, 121, 4280–4286.
- Komrokji, R.S. Current state of the art: Management of higher risk myelodysplastic syndromes. Clin. Lymphoma Myeloma Leuk. 2016, 16, S39–S43.
- Garcia-Manero, G. Myelodysplastic syndromes: 2015 Update on diagnosis, risk-stratification and management. Am. J. Hematol. 2015, 90, 831–841.
- Malcovati, L.; Hellström-Lindberg, E.; Bowen, D.; Adès, L.; Cermak, J.; Del Cañizo, C.; Della Porta, M.G.; Fenaux, P.; Gattermann, N.; Germing, U.; et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: Recommendations from the European LeukemiaNet. Blood 2013, 122, 2943–2964.
- Stojkov, K.; Silzle, T.; Stussi, G.; Schwappach, D.; Bernhard, J.; Bowen, D.; Čermák, J.; Dinmohamed, A.G.; Eeltink, C.; Eggmann, S.; et al. Guideline-based indicators for adult patients with myelodysplastic syndromes. Blood Adv. 2020, 4, 4029–4044.
- Kipfer, B.; Daikeler, T.; Kuchen, S.; Hallal, M.; Andina, N.; Allam, R.; Bonadies, N. Increased cardiovascular comorbidities in patients with myelodysplastic syndromes and chronic myelomonocytic leukemia presenting with systemic inflammatory and autoimmune manifestations. Semin. Hematol. 2018, 55, 242–247.
- Fenaux, P.; Platzbecker, U.; Ades, L. How we manage adults with myelodysplastic syndrome. Br. J. Haematol. 2020, 189, 1016–1027.
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506.
- Malcovati, L.; Papaemmanuil, E.; Ambaglio, I.; Elena, C.; Gallì, A.; Della Porta, M.G.; Travaglino, E.; Pietra, D.; Pascutto, C.; Ubezio, M.; et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood 2014, 124, 1513–1521.
- Bejar, R.; Stevenson, K.E.; Caughey, B.; Lindsley, C.; Mar, B.; Stojanov, P.; Getz, G.; Steensma, D.P.; Ritz, J.; Soiffer, R.; et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J. Clin. Oncol. 2014, 32, 2691–2698.
- Chanias, I.; Bonadies, N. Current Standard of Care in Patients with Myelodysplastic Syndromes and Future Perspectives. Heal. Online Med. J. 2020, 10–22.
More
Information
Subjects:
Pathology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
3 times
(View History)
Update Date:
22 Sep 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No