 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sandra Berndt | + 1074 word(s) | 1074 | 2021-06-21 11:08:48 | | | |
| 2 | Camila Xu | + 242 word(s) | 1316 | 2021-07-14 07:59:44 | | |
Video Upload Options
Src family kinases (SFKs) are key regulators of cell proliferation, differentiation, and survival. The expression of these non-receptor tyrosine kinases is strongly correlated with cancer development and tumor progression. Thus, this family of proteins serves as an attractive drug target.
1. Introduction
Src family kinases (SFKs) are non-receptor protein-tyrosine kinases that regulate essential processes such as cell proliferation, differentiation, survival, migration, and metabolism [1].
SFKs are upregulated in malignancies, and their expression levels as well as specific activity are elevated in brain, breast, colon, lung, and pancreatic carcinomas [2][3][4][5][6][7][8][9]. For acute myeloid leukemia and colorectal cancer, a direct correlation between expression level of some SFK family members and patient survival was observed [10][11].
The human SFKs consist of eight typical family members (Src, Fyn, Yes, Fgr, Hck, Blk, Lck, and Lyn) and three atypical family members (Brk, Frk, Srms) based on sequence similarity. In some nomenclatures, atypical family members are considered a separate family also called the Frk family [12][13]. Most of the SFKs, such as Yes, Fgr, Blk, Hck, Lck, and Lyn have an important regulatory role in signaling pathways of hematopoietic cells. A majority of SFKs are essential in immune response; whereas Fyn and Lck are activated immediately after T-cell receptor stimulation, expression of Fgr, Hck, Lyn is induced in stimulated mature monocytes and macrophages [14].
SFKs represent drug targets with great therapeutic potential, especially in cancer treatment, since SFKs are involved in cancer progression in various stages (reviewed in [15][16]). Approved drugs for cancer treatment targeting Src family kinases, such as Dasatinib, show a high level of toxicity [17] due to their unselective inhibition of SFKs in cancer and healthy cells. In-depth structural, functional, and mechanistic knowledge of each single SFK in combination with a detailed understanding of their expression regulation can be the basis for a more specific therapeutic approach with limited side effects.
2. Activation mechanism of SFKs
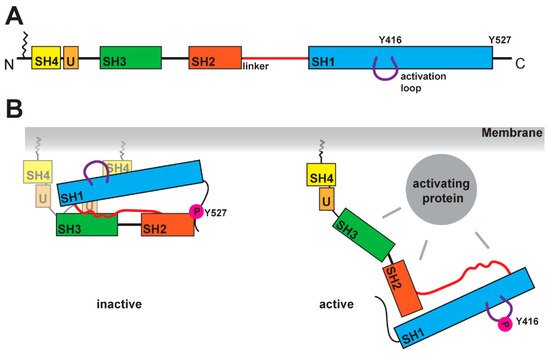
SFKs are composed of distinct domains (Figure 1A). The N-terminal region, also called the SH4 domain, contains a myristoylation or palmitoylation site, which acts as a membrane anchor and is a key element for the localization of SFKs [19,20]. The unique domain, which is located after the SH4 domain, has a regulatory function for membrane localization and can form a fuzzy intramolecular complex with the neighboring SH3 domain [21,22,23]. SH3 domains serve as binding elements and are known to interact with a variety of polyproline motifs (reviewed in [24]). After a linker, SFKs contain the SH2 domain, known to interact with phosphorylated tyrosine residues, and following a longer linker region, the kinase domain, containing an N-lobe and a C-lobe. This domain entails two regulatory phosphorylation sites (Y-416 and Y-527 for Src as a representative example, Figure 1B) [25,26]. The first regulatory site is the activating autophosphorylation site, and the second one the negative regulatory site. The phosphorylation and dephosphorylation of these tyrosine residues cause dramatic structural changes and affect the activity of the kinase. In the inactive structure, Y-527 is phosphorylated by CSK (C-terminal Src kinase) or CHK (CSK homologous kinase) [27,28], which results in an interaction of the kinase domain with the SH2 domain [25,29,30]. This inactive conformation is further stabilized by the binding of the SH3 domain with the polyproline motif in the linker region between SH2 domain and kinase domain [30,31]. A recent finding showed a possible involvement of the SH4 domain, which binds in the inactive conformation to the kinase domain [32]. This compact state results in a closed conformation of the N- and C-lobes in the kinase domain, which results in a shielding of the Y-416 in the active site. In this closed conformation, the binding of ATP and substrates is blocked. In the active conformation, the interactions of SH2 and SH3 domains are displaced by other binding partners, which results in an open conformation (Figure 1B). This grants accessibility of the active site and allows for autophosphorylation of Y-416.
In general, SFKs are activated by several different growth factor receptor tyrosine kinases. For example, the SH2 domains interact with SHP-1 protein tyrosine phosphatase, CRK-associated substrate, or protein tyrosine phosphatase-1B [18][19][20]. Proteins with typical polyproline motifs such as cyclin-dependent kinase-5, KCNB1,
Additionally, for a number of G protein-coupled receptors (GPCRs), SFK activation was shown. However, the exact activation mechanism of this interaction is poorly understood. It has been postulated that there are three ways of GPCR-mediated SFK activation: through arrestins, G proteins, or direct binding. A detailed understanding of the mechanism is highly desirable due to the potential druggability of GPCRs and the crucial role of SFKs in cancer development and progression.
3. Src Activation through a GPCR–Arrestin Complex
Until now, the best understood GPCR-mediated activation of SFK is arrestin-based. As early as 1999, arrestin-2-mediated Src activation by beta-2 adrenergic receptor (β2AR) stimulation was detected [21]. Later on, this was observed for multiple other receptors. Until recently, there was no evidence of how this interaction could take place.
Nevertheless, in the ‘tail’ conformation, arrestin internalization and signaling are still possible. The most dominant conformational change during the activation of arrestin is the rotation of the N- and C-domains towards each other. With this domain rotation, multiple small conformational changes appear (also called switch regions for arrestin-3) The interaction of SFK SH3 domains with polyproline motifs in arrestin are substantial for the activation of SFKs [22].
Yang et al showed that the receptor phospho-tail allosterically regulates the different conformations within the polyproline motifs in arrestin-2, which subsequently allows for the binding of the SFK SH3 domain. leading to the adoption of an open active conformation of the kinase [22]. There are only a few activation studies for arrestin-3-mediated Src activation. For dopamine D1 receptor, activation of Src in the presence of arrestin-3 was shown [23].
4. Src Activation by G Proteins
G proteins (heterotrimeric guanine nucleotide-binding regulatory proteins) contain α, ß, and γ subunits. Agonist-bound GPCRs activate G proteins by facilitating the exchange of GDP to GTP at the α subunit. Activation of SFKs by G proteins can be achieved through either the α subunit or the ßγ subunit. The described two switch regions in G proteins are defined regions crucial for binding of effectors such as Ras protein or adenylyl cyclase [24][25][26].
The two sites of phosphorylation are Y37 and Y377 [27][28], and both promote GTP hydrolysis. The different regulatory mechanisms by G protein phosphorylation are reviewed elsewhere [29]. In transducin, an additional phosphorylation site, Y142, was found [30]. Overall, an arrestin-independent G protein-mediated activation of Src is still not fully understood and requires further investigation.
References
- Tice, D.A.; Biscardi, J.S.; Nickles, A.L.; Parsons, S.J. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1415–1420.
- Cartwright, C.A.; Kamps, M.P.; Meisler, A.I.; Pipas, J.M.; Eckhart, W. pp60c-src activation in human colon carcinoma. J. Clin. Investig. 1989, 83, 2025–2033.
- Talamonti, M.S.; Roh, M.S.; Curley, S.A.; Gallick, G.E. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J. Clin. Investig. 1993, 91, 53–60.
- Guarino, M. Src signaling in cancer invasion. J. Cell. Physiol. 2010, 223, 14–26.
- Luttrell, D.K.; Lee, A.; Lansing, T.J.; Crosby, R.M.; Jung, K.D.; Willard, D.; Luther, M.; Rodriguez, M.; Berman, J.; Gilmer, T.M. Involvement of pp60c-src with two major signaling pathways in human breast cancer. Proc. Natl. Acad. Sci. USA 1994, 91, 83–87.
- Garmendia, I.; Pajares, M.J.; Hermida-Prado, F.; Ajona, D.; Bértolo, C.; Sainz, C.; Lavín, A.; Remírez, A.B.; Valencia, K.; Moreno, H.; et al. YES1 Drives Lung Cancer Growth and Progression and Predicts Sensitivity to Dasatinib. Am. J. Respir. Crit. Care Med. 2019, 200, 888–899.
- Staley, C.A.; Parikh, N.U.; Gallick, G.E. Decreased tumorigenicity of a human colon adenocarcinoma cell line by an antisense expression vector specific for c-Src. Cell Growth Differ. 1997, 8, 269–274.
- Stettner, M.R.; Wang, W.; Nabors, L.B.; Bharara, S.; Flynn, D.C.; Grammer, J.R.; Gillespie, G.Y.; Gladson, C.L. Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res. 2005, 65, 5535–5543.
- Alcalá, S.; Mayoral-Varo, V.; Ruiz-Cañas, L.; López-Gil, J.C.; Heeschen, C.; Martín-Pérez, J.; Sainz, B. Targeting SRC Kinase Signaling in Pancreatic Cancer Stem Cells. Int. J. Mol. Sci. 2020, 21.
- Patel, R.K.; Weir, M.C.; Shen, K.; Snyder, D.; Cooper, V.S.; Smithgall, T.E. Expression of myeloid Src-family kinases is associated with poor prognosis in AML and influences Flt3-ITD kinase inhibitor acquired resistance. PLoS ONE 2019, 14, e0225887.
- Han, N.M.; Curley, S.A.; Gallick, G.E. Differential activation of pp60(c-src) and pp62(c-yes) in human colorectal carcinoma liver metastases. Clin. Cancer Res. 1996, 2, 1397–1404.
- Qiu, H.; Miller, W.T. Regulation of the nonreceptor tyrosine kinase Brk by autophosphorylation and by autoinhibition. J. Biol. Chem. 2002, 277, 34634–34641.
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557.
- Kovács, M.; Németh, T.; Jakus, Z.; Sitaru, C.; Simon, E.; Futosi, K.; Botz, B.; Helyes, Z.; Lowell, C.A.; Mócsai, A. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J. Exp. Med. 2014, 211, 1993–2011.
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358.
- Biscardi, J.S.; Tice, D.A.; Parsons, S.J. c-Src, receptor tyrosine kinases, and human cancer. Adv. Cancer Res. 1999, 76, 61–119.
- Brunner, A.M.; Costa, D.B.; Heist, R.S.; Garcia, E.; Lindeman, N.I.; Sholl, L.M.; Oxnard, G.R.; Johnson, B.E.; Hammerman, P.S. Treatment-related toxicities in a phase II trial of dasatinib in patients with squamous cell carcinoma of the lung. J. Thorac. Oncol. 2013, 8, 1434–1437.
- Somani, A.K.; Bignon, J.S.; Mills, G.B.; Siminovitch, K.A.; Branch, D.R. Src kinase activity is regulated by the SHP-1 protein-tyrosine phosphatase. J. Biol. Chem. 1997, 272, 21113–21119.
- Goldberg, G.S.; Alexander, D.B.; Pellicena, P.; Zhang, Z.-Y.; Tsuda, H.; Miller, W.T. Src phosphorylates Cas on tyrosine 253 to promote migration of transformed cells. J. Biol. Chem. 2003, 278, 46533–46540.
- Bjorge, J.D.; Pang, A.; Fujita, D.J. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J. Biol. Chem. 2000, 275, 41439–41446.
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 1999, 283, 655–661.
- Yang, F.; Xiao, P.; Qu, C.-X.; Liu, Q.; Wang, L.-Y.; Liu, Z.-X.; He, Q.-T.; Liu, C.; Xu, J.-Y.; Li, R.-R.; et al. Allosteric mechanisms underlie GPCR signaling to SH3-domain proteins through arrestin. Nat. Chem. Biol. 2018, 14, 876–886.
- Kaya, A.I.; Perry, N.A.; Gurevich, V.V.; Iverson, T.M. Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 14139–14149.
- Berlot, C.H.; Bourne, H.R. Identification of effector-activating residues of Gs alpha. Cell 1992, 68, 911–922.
- Tesmer, J.J.; Sunahara, R.K.; Gilman, A.G.; Sprang, S.R. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha.GTPgammaS. Science 1997, 278, 1907–1916.
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.-Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868.
- Moyers, J.S.; Linder, M.E.; Shannon, J.D.; Parsons, S.J. Identification of the in vitro phosphorylation sites on Gs alpha mediated by pp60c-src. Biochem. J. 1995, 305, 411–417.
- Hausdorff, W.P.; Pitcher, J.A.; Luttrell, D.K.; Linder, M.E.; Kurose, H.; Parsons, S.J.; Caron, M.G.; Lefkowitz, R.J. Tyrosine phosphorylation of G protein alpha subunits by pp60c-src. Proc. Natl. Acad. Sci. USA 1992, 89, 5720–5724.
- Chakravorty, D.; Assmann, S.M. G protein subunit phosphorylation as a regulatory mechanism in heterotrimeric G protein signaling in mammals, yeast, and plants. Biochem. J. 2018, 475, 3331–3357.
- Bell, M.W.; Desai, N.; Guo, X.X.; Ghalayini, A.J. Tyrosine phosphorylation of the alpha subunit of transducin and its association with Src in photoreceptor rod outer segments. J. Neurochem. 2000, 75, 2006–2019.


