Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jin-Ho Koh | + 2028 word(s) | 2028 | 2021-05-25 08:59:17 | | | |
| 2 | Peter Tang | Meta information modification | 2028 | 2021-07-09 08:13:26 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Koh, J. PGC-1α in Metabolic Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/11862 (accessed on 07 February 2026).
Koh J. PGC-1α in Metabolic Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/11862. Accessed February 07, 2026.
Koh, Jin-Ho. "PGC-1α in Metabolic Diseases" Encyclopedia, https://encyclopedia.pub/entry/11862 (accessed February 07, 2026).
Koh, J. (2021, July 09). PGC-1α in Metabolic Diseases. In Encyclopedia. https://encyclopedia.pub/entry/11862
Koh, Jin-Ho. "PGC-1α in Metabolic Diseases." Encyclopedia. Web. 09 July, 2021.
Copy Citation
Mitochondria contain the majority of cellular nicotinamide adenine dinucleotide (NAD+), which an essential cofactor that regulates metabolic function. A decrease in both mitochondria biogenesis and NAD+ is a characteristic of metabolic diseases, and peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) orchestrates mitochondrial biogenesis and is involved in mitochondrial NAD+ pool.
mitochondria
PGC-1α
NAD+
SIRTs
metabolic disease
1. Introduction
Mitochondria are powerhouses that generate the majority of cellular ATP via fatty acid oxidation, tricarboxylic acid (TCA) cycle, electron transport chain (ETC), and ATP synthase. Mitochondrial dysfunction is linked to metabolic diseases and health issues, including insulin resistance and type 2 diabetes, cancer, Alzheimer’s disease, and others [1][2][3].
Nicotinamide adenine dinucleotide (NAD+) is an essential cofactor that regulates metabolic function, and it is an electron carrier and signaling molecule involved in response to alterations in the cellular metabolic redox state, including muscle contraction, high-fat diet (HFD), insulin resistance, and type 2 diabetes mellitus (T2DM) [4]. NAD+ plays a key role in cellular signaling and regulation of metabolism in glycolysis, oxidative phosphorylation, the TCA cycle, and DNA repair [5][6][7][8]. Moreover, NAD+ is reduced to NADH by accepting two electrons and a proton from glycolysis and the TCA cycle, and mitochondrial NADH is oxidized through mitochondrial respiratory complex I (NADH ubiquinone oxidoreductase) in the ETC [9]. This is one of the essential steps during oxidative phosphorylation; therefore, an optimal ratio of NAD+/NADH is required for mitochondrial metabolism [10][11][12], and lower NAD+ levels and dysregulation of NAD+/NADH ratio can be one of the reasons for developing metabolic diseases and T2DM [9]. In particular, NAD+ biosynthesis and its function crucially influence the bioenergetic process in mitochondria [13] and are clearly linked to peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) [14][15][16][17][18].
2. NAD+–SIRT1–PGC-1α Pathway in Metabolic Diseases
Plasma glucose homeostasis is critical for the functioning of mammalian organisms; thus, glucose levels should be strictly regulated according to nutrient conditions and energy demands. To maintain glucose homeostasis at the cellular level and to adapt to various challenges such as high-nutrient condition, disuse, and sarcopenia, it is necessary to improve or stabilize mitochondrial function, number, and size, which are important for maintaining the cellular NAD+ pool [16][19][20][21][22][23].
PGC-1α is a master regulator that interacts with various transcription factors involved in cellular metabolic functions [24]; thus, PGC-1α mediates the transcriptional activity and biological response related to them [24]. SIRT1 (Sirtuin 1) is involved in the regulation of systemic metabolism via the control of glucose and lipid homeostasis by deacetylating various targets, especially PGC-1α [25]. Therefore, the NAD+–SIRTs–PGC-1α pathway plays a vital role in cellular metabolic function. NAD+ depletion is a characteristic of diabetes [26], and sirtuins, including SIRT1-3 and SIRT6, influence cellular functions such as glucose metabolism, mitochondrial function, and oxidative stress [25][27][28][29]. It is well documented that PGC-1α expression is reduced in T2DM muscle [30][31][32].
The NAD+ pool is important for cell physiological and metabolic functions for cell integrity; however, metabolic diseases, such as insulin resistance in tissues and diabetes, increase NAD+ consumption; thus, lower levels of cellular NAD+ are clearly linked to metabolic diseases [33][34][35][36][37][38][39]. In this context, SIRT1, which consumes NAD+ for cellular metabolic function, is downregulated in several cells and tissues, including myotubes, HEK293, peripheral blood mononuclear cell, human skeletal muscle, and adipose tissue, in insulin-resistant states [39][40][41]. A previous study has shown that SIRT1 regulates glucose homeostasis by regulating the secretion of insulin and protecting beta (β)-cells in the pancreas [42], enhancing mitochondrial biogenesis and glucose uptake in skeletal muscle [43], and promoting glucose production and fatty acid oxidation in the liver [44]. β-cell-specific SIRT1 overexpression in mice improves insulin secretion and glucose tolerance in response to glucose [42]. Age-related downregulation of SIRT1 activity due to a lack of systemic NAD+ biosynthesis results in a decrease in insulin secretion from β-cells in response to glucose; however, treatment with nicotinamide mononucleotide (NMN), which is a derivative of niacin and an intermediate in NAD+ biosynthesis in the salvage pathway, restores insulin secretion and improves glucose tolerance in aged mice with β-cell-specific SIRT1 overexpression. Therefore, SIRTs regulate glucose–lipid metabolism and mitochondrial biogenesis via PGC-1α [42][43][44][45]. Overall, NAD+ boosting can be one of the strategies to improve metabolic dysfunction via SIRTs–PGC-1α; therefore, we will discuss the role of the SIRTs–PGC-1α pathway in increasing NAD+ biosynthesis and decreasing NAD+ consumption.
3. NAD+ Biosynthesis
Cellular NAD+ availability is maintained by the regulation of NAD+ biosynthesis and degradation. There are five major precursors and intermediates in NAD+ synthesis in mammals; tryptophan (Trp), nicotinamide (NAM), nicotinic acid (NA), nicotinamide riboside (NR), and nicotinamide mononucleotide (NMN); they stimulate NAD+ synthesis via different pathways [46] (Figure 1). These pathways can synthesize 300–800 µM of cellular NAD+, depending upon the tissue and organ [47][48][49][50]. In addition, NAD+ can be resynthesized from an intermediate, such as NMN, of NAD+. This section focuses on the NAD+ biosynthesis pathway.
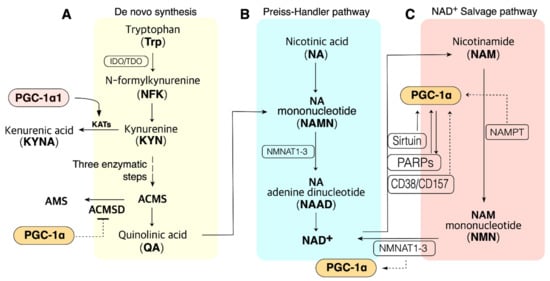
Figure 1. PGC-1α is involved in the pathway of NAD+ biosynthesis and consumption in the metabolic tissue such as muscle, liver, and adipose tissue. (A). NAD+ de novo synthesis from Trp. PGC-1α may promote QA synthesis via blocking ACMSD. PGC-1α1 may promote de novo synthesis via KAT activation. (B). NAD+ biosynthesis from NA in the Preiss–Handler pathway; QA from de novo synthesis also can be synthesized to NAD+ in the Preiss–Handler pathway. (C). PGC-1α may influence many enzymes in the salvage pathway. ACMS, α-amino-β-carboxymuconate-ε-semialdehyde; ACMSD, ACMS decarboxylase; NAMPT, nicotinamide phosphoribosyltransferases (three isoforms exist); NMNAT, NMN adenylyltransferase; PARP, poly (ADP-ribose) polymerase. Dashed lines indicate additional evidence is required to reveal the mechanisms.
4. PGC-1α1 Regulates the Mitochondrial NAD+ Pool via Malate–Aspartate Shuttle
NAD+ levels in mitochondria are important for maintaining metabolic functions and cell survival in oxidative metabolic tissues, including the skeletal muscle, heart, and liver. In metabolic tissues that dominantly use oxidative phosphorylation to generate ATP, NAD+ levels in mitochondria should be maintained at a higher level than that in the cytoplasm [13][51][52][53]. Malate dehydrogenase (MDH) and aspartate aminotransferase (AST) form the malate–aspartate shuttle (MAS), which plays a vital role in the exchange of cytosolic NADH for mitochondrial NAD+, which is an irreversible step in the exchange of mitochondrial aspartate and cytosolic glutamate and a proton by the aspartate–glutamate carrier (AGC) [54][55][56] (Figure 2). Thus, MAS can regulate mitochondrial NAD+ pool, and since the mitochondrial NAD+ pool is well maintained and higher than that in the cytoplasm (cytosolic/nuclear NAD+ levels are ~100 µM, while mitochondrial NAD+ levels are ~250 µM) [53], mitochondrial numbers and size regulate NAD+ levels in metabolic tissue. Mitochondria function as an NAD+ warehouse. Even if a large amount of NAD+ is depleted from the cytoplasm, mitochondrial NAD+ levels can be conserved for at least 24 h and possibly up to three days [47][48][51][57][58][59], indicating that mitochondrial NAD+ has specific roles in metabolism and is separated from the cytoplasm.
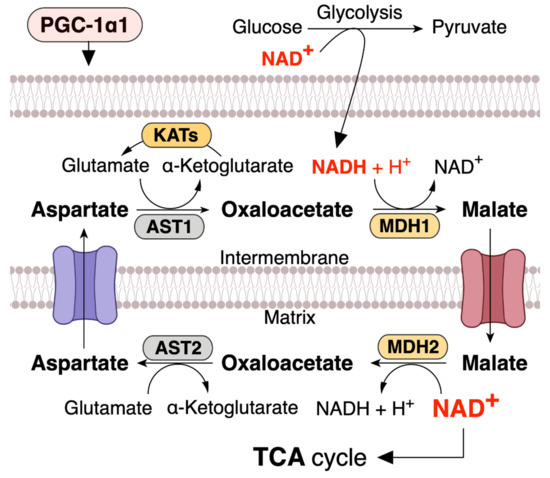
Figure 2. PGC-1α1 regulates malate and aspartate system (MAS) and increases mitochondrial NAD+ levels in skeletal muscle. NADH produced during glycolysis enters the mitochondrial matrix via the MAS. MDH, malate dehydrogenase; AST, aspartate aminotransferase; KATs, kynurenine aminotransferases. This concept was referenced from the study by Agudelo et al. (2019) [60].
This speculation leads to the hypothesis that MAS-induced increase in the mitochondrial NAD+ pool can increase the expression of sirtuin family (SIRT1-7) members and PGC-1α. Indeed, a previous study has shown that MAS regulates the intracellular NAD+/NADH ratio; moreover, calorie restriction increases the mitochondrial NAD+ pool and SIRT2 expression via MAS [54].
An AGC1 knockout study has been shown to decrease the cellular NAD+/NADH ratio and impair aspartate delivery to the cytosol [61]. Moreover, muscle-specific PGC-1α1 overexpression in mice enhances the expression of SLC25A12, another gene name for AGC1 [60]. MAS activated by PGC-1α1 rigidly maintains the NAD+ pool to maintain oxidative metabolism when energy demand is high (Figure 2), such as muscle contraction during exercise. Trained muscle has a higher level of NAD+ pool [20], and PGC-1α1 regulates mitochondrial biogenesis and cellular oxidative metabolism. Overall, these studies lead us to speculate that MAS is linked to mitochondrial and metabolic functions. Furthermore, a previous study has shown that deficiency in MDH, which plays an essential role in the MAS and TCA cycle, is a metabolic defect characterized by a severe neurodevelopmental phenotype [62].
5. Role of PGC-1α in NAD+ Metabolism in Metabolic Diseases
SIRT1 is a nicotinamide adenosine dinucleotide (NAD)-dependent deacetylase that removes acetyl groups from histone and nonhistone proteins [63]. It potentially mediates the effects of calorie restriction on health benefits for longevity [64], and exercise also increases NAD+/NADH turnover [65]. Increased NAD+/NADH turnover rate by glycolysis, TCA cycle, and mitochondrial oxidative phosphorylation system during cellular high energy-demand and increased NAD+ by SIRT1 increase mitochondrial biogenesis [4], which induces the salvage pathway to regenerate NAD+, because this pathway can quickly regenerate NAD+ from NAM in two steps. NAMPT and NMNAT are enzymes involved in the salvage pathway.
The findings to date show that PGC-1α has the most influence on the salvage mechanism of NAD+ metabolism. PGC-1α is activated by specific SIRTs, and PGC-1α promotes NAD+ re-biosynthesis via the salvage pathway and increases mitochondrial biogenesis, thereby improving mitochondrial function and protecting against high fat diet induced obesity [66]. Here, we focus on the PGC-1α mechanism in various NAD+ consumption and biosynthesis pathways related to metabolic diseases.
6. NAD+–SIRT1–PGC-1α Pathway in Diabetes
Mitochondrial function is involved in whole-body and cellular glucose homeostasis, and mitochondrial functions are decreased in states of insulin resistance and diabetes [2]. It is well established that an increase in mitochondrial function by exercise training in various metabolic tissue, including skeletal muscle and adipose tissue, of the insulin resistance or diabetes subjects improves glucose homeostasis [67][68]. Thus, mitochondrial function is important for glucose homeostasis.
SIRTs directly interact with PGC-1α and deacetylate at specific lysine residues in an NAD+-dependent manner, activating PGC-1α [69]. Thus, NAD+–SIRTs–PGC-1α pathway plays a critical role in glucose homeostasis in diabetes and lifespan [69]. As we discussed above, since mitochondria function as a warehouse of NAD+, and PGC-1α regulates mitochondrial biogenesis in metabolic tissue, NAD+, SIRT1, and PGC-1α play an important role in their function of regulating glucose homeostasis in each other. In metabolic tissue, including skeletal muscle, liver, and adipose tissue, of T2DM state, PGC-1α protein and NAD+ levels are downregulated, as is SIRT1 activity [33][70][71][72]. These shifts are clearly linked to the mitochondrial functions, numbers, and size [33][70][71][72].
As the strategy to increase cellular NAD+ levels, the biosynthesis of NAD+ should be increased, and its consumption should be prevented. Daily intake of NAD+ precursor is one of the strategies to increase NAD+ synthesis and improve metabolic function in diabetes. Indeed, a previous study has shown that tryptophan supplementation increases lifespan through NAD+ de novo synthesis [73], and acipimox, an NAD+ precursor, can directly improve skeletal muscle mitochondrial function in T2DM patients [74]. Mice receiving NR were protected against high fat diet induced weight gain and had higher insulin sensitivity with increased mitochondrial content in skeletal muscle and brown adipose tissue [75]. Niacin supplementation can improve lipid profiles in T2DM patients [76]. These data suggest that supplementation of NAD+ precursor can be one of the strategies to improve insulin resistance and treat T2DM (Figure 3).
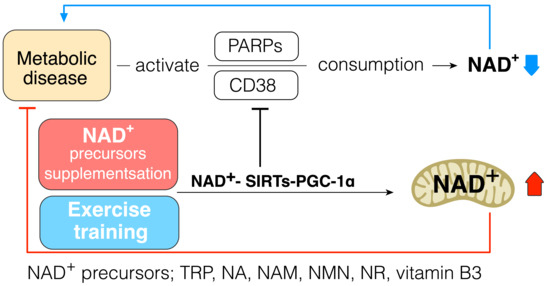
Figure 3. NAD+–SIRTs–PGC-1α pathway increases NAD+ pool and inhibits NAD+ consumption. NAD+ precursors: tryptophan (Trp), nicotinic acid (NA), nicotinamide (NAM), NAM mononucleotide (NMN), nicotinamide riboside (NR), vitamin B3; metabolic diseases: insulin resistance, type 2 diabetes.
It is widely accepted that exercise can provide many health-beneficial effects for T2DM patients; in this context, exercise could increase the cellular NAD+ pool in an indirect way via endogenous enzyme alteration to activate its synthesis or protect the NAD+ pool against overconsumption. Indeed, a previous study has revealed that trained muscle has a higher level of NAD+ pool, and aerobic and resistance exercise training improves the capacity of NAD+ salvage pathway in aged human skeletal muscle [20]. AMPK is activated by exercise [77], and it is well evidenced that AMPK regulates the gene expression related to energy metabolism in mouse skeletal muscle via coordination with SIRT1 and enhances its activity by increasing cellular NAD+ pool [78]. Exercise increases NAMPT in human skeletal muscle [79], and NAMPT overexpression in mice skeletal muscle increases muscle NAD+ levels and protects against body weight gain in high-fat-fed mice, as well as increasing mitochondrial gene expression and endurance capacity [80]. Exercise-training-induced increase in mitochondrial biogenesis in tissue also protects against overconsumption of NAD+ because the mitochondrial capacity for NAD+ conservation is higher than that of the cytoplasm [47][48][51][57][58][59] (Figure 3).
References
- Kristian, T.; Balan, I.; Schuh, R.; Onken, M. Mitochondrial dysfunction and nicotinamide dinucleotide catabolism as mechanisms of cell death and promising targets for neuroprotection. J. Neurosci. Res. 2011, 89, 1946–1955.
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103.
- Solaini, G.; Sgarbi, G.; Baracca, A. Oxidative phosphorylation in cancer cells. Biochim. Biophys. Acta 2011, 1807, 534–542.
- Navas, L.E.; Carnero, A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2.
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19.
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53.
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; de Giorgis, T.; Kiess, W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015, 11, 535–546.
- Johnson, S.; Imai, S.I. NAD (+) biosynthesis, aging, and disease. F1000Res 2018, 7, 132.
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31.
- Pittelli, M.; Felici, R.; Pitozzi, V.; Giovannelli, L.; Bigagli, E.; Cialdai, F.; Romano, G.; Moroni, F.; Chiarugi, A. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharmacol. 2011, 80, 1136–1146.
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468.
- Alano, C.C.; Ying, W.; Swanson, R.A. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J. Biol. Chem. 2004, 279, 18895–18902.
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428.
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxidative Med. Cell. Longev. 2020, 2020, 1452696.
- Chandrasekaran, K.; Anjaneyulu, M.; Choi, J.; Kumar, P.; Salimian, M.; Ho, C.Y.; Russell, J.W. Role of mitochondria in diabetic peripheral neuropathy: Influencing the NAD(+)-dependent SIRT1-PGC-1α-TFAM pathway. Int. Rev. Neurobiol. 2019, 145, 177–209.
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105.
- Adamovich, Y.; Shlomai, A.; Tsvetkov, P.; Umansky, K.B.; Reuven, N.; Estall, J.L.; Spiegelman, B.M.; Shaul, Y. The protein level of PGC-1α, a key metabolic regulator, is controlled by NADH-NQO1. Mol. Cell Biol. 2013, 33, 2603–2613.
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532.
- Lawler, J.M.; Hindle, A. Living in a box or call of the wild? Revisiting lifetime inactivity and sarcopenia. Antioxid. Redox Signal. 2011, 15, 2529–2541.
- de Guia, R.M.; Agerholm, M.; Nielsen, T.S.; Consitt, L.A.; Sogaard, D.; Helge, J.W.; Larsen, S.; Brandauer, J.; Houmard, J.A.; Treebak, J.T. Aerobic and resistance exercise training reverses age-dependent decline in NAD(+) salvage capacity in human skeletal muscle. Physiol. Rep. 2019, 7, e14139.
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194.
- Zorzano, A.; Hernandez-Alvarez, M.I.; Palacin, M.; Mingrone, G. Alterations in the mitochondrial regulatory pathways constituted by the nuclear co-factors PGC-1alpha or PGC-1beta and mitofusin 2 in skeletal muscle in type 2 diabetes. Biochim. Biophys. Acta 2010, 1797, 1028–1033.
- Ritov, V.B.; Menshikova, E.V.; Azuma, K.; Wood, R.; Toledo, F.G.; Goodpaster, B.H.; Ruderman, N.B.; Kelley, D.E. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E49–E58.
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839.
- Kitada, M.; Kume, S.; Kanasaki, K.; Takeda-Watanabe, A.; Koya, D. Sirtuins as possible drug targets in type 2 diabetes. Curr. Drug Targets 2013, 14, 622–636.
- Garcia Soriano, F.; Virág, L.; Jagtap, P.; Szabó, E.; Mabley, J.G.; Liaudet, L.; Marton, A.; Hoyt, D.G.; Murthy, K.G.; Salzman, A.L.; et al. Diabetic endothelial dysfunction: The role of poly(ADP-ribose) polymerase activation. Nat. Med. 2001, 7, 108–113.
- Gomes, P.; Fleming Outeiro, T.; Cavadas, C. Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism. Trends Pharmacol. Sci. 2015, 36, 756–768.
- McDonnell, E.; Peterson, B.S.; Bomze, H.M.; Hirschey, M.D. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol. Metab. 2015, 26, 486–492.
- Kuang, J.; Chen, L.; Tang, Q.; Zhang, J.; Li, Y.; He, J. The Role of Sirt6 in Obesity and Diabetes. Front. Physiol. 2018, 9, 135.
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273.
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471.
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671.
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536.
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. NNMT activation can contribute to the development of fatty liver disease by modulating the NAD (+) metabolism. Sci. Rep. 2018, 8, 8637.
- Yang, X.; Liu, Q.; Li, Y.; Tang, Q.; Wu, T.; Chen, L.; Pu, S.; Zhao, Y.; Zhang, G.; Huang, C.; et al. The diabetes medication canagliflozin promotes mitochondrial remodelling of adipocyte via the AMPK-Sirt1-Pgc-1alpha signalling pathway. Adipocyte 2020, 9, 484–494.
- Wang, S.; Wan, T.; Ye, M.; Qiu, Y.; Pei, L.; Jiang, R.; Pang, N.; Huang, Y.; Liang, B.; Ling, W.; et al. Nicotinamide riboside attenuates alcohol induced liver injuries via activation of SirT1/PGC-1α/mitochondrial biosynthesis pathway. Redox Biol. 2018, 17, 89–98.
- Wang, P.; Zhang, R.Y.; Song, J.; Guan, Y.F.; Xu, T.Y.; Du, H.; Viollet, B.; Miao, C.Y. Loss of AMP-activated protein kinase-alpha2 impairs the insulin-sensitizing effect of calorie restriction in skeletal muscle. Diabetes 2012, 61, 1051–1061.
- Waldman, M.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Kornwoski, R.; Aravot, D.; Arad, M.; Hochhauser, E. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1α axis. Exp. Cell Res. 2018, 373, 112–118.
- de Kreutzenberg, S.V.; Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Semplicini, A.; Dalla Man, C.; Cobelli, C.; Fadini, G.P.; Avogaro, A. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: Potential biochemical mechanisms. Diabetes 2010, 59, 1006–1015.
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245.
- Fröjdö, S.; Durand, C.; Molin, L.; Carey, A.L.; El-Osta, A.; Kingwell, B.A.; Febbraio, M.A.; Solari, F.; Vidal, H.; Pirola, L. Phosphoinositide 3-kinase as a novel functional target for the regulation of the insulin signaling pathway by SIRT1. Mol. Cell Endocrinol. 2011, 335, 166–176.
- Moynihan, K.A.; Grimm, A.A.; Plueger, M.M.; Bernal-Mizrachi, E.; Ford, E.; Cras-Méneur, C.; Permutt, M.A.; Imai, S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005, 2, 105–117.
- Zhang, H.H.; Ma, X.J.; Wu, L.N.; Zhao, Y.Y.; Zhang, P.Y.; Zhang, Y.H.; Shao, M.W.; Liu, F.; Li, F.; Qin, G.J. SIRT1 attenuates high glucose-induced insulin resistance via reducing mitochondrial dysfunction in skeletal muscle cells. Exp. Biol. Med. (Maywood) 2015, 240, 557–565.
- Rodgers, J.T.; Puigserver, P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc. Natl. Acad. Sci. USA 2007, 104, 12861–12866.
- Guarente, L.; Franklin, H. Epstein Lecture: Sirtuins, aging, and medicine. N. Engl. J. Med. 2011, 364, 2235–2244.
- Imai, S. Nicotinamide phosphoribosyltransferase (Nampt): A link between NAD biology, metabolism, and diseases. Curr. Pharm. Des. 2009, 15, 20–28.
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107.
- Houtkooper, R.H.; Cantó, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223.
- Wilhelm, F.; Hirrlinger, J. The NAD+ /NADH redox state in astrocytes: Independent control of the NAD+ and NADH content. J. Neurosci. Res. 2011, 89, 1956–1964.
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Hashimoto, T.; Tsuchiya, M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J. Biol. Chem. 2007, 282, 24574–24582.
- Di Lisa, F.; Menabò, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 2001, 276, 2571–2575.
- Sauve, A.A. NAD+ and vitamin B3: From metabolism to therapies. J. Pharmacol. Exp. Ther. 2008, 324, 883–893.
- Cambronne, X.A.; Stewart, M.L.; Kim, D.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. Biosensor reveals multiple sources for mitochondrial NAD⁺. Science 2016, 352, 1474–1477.
- Easlon, E.; Tsang, F.; Skinner, C.; Wang, C.; Lin, S.J. The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev. 2008, 22, 931–944.
- Greenhouse, W.V.; Lehninger, A.L. Occurrence of the malate-aspartate shuttle in various tumor types. Cancer Res. 1976, 36, 1392–1396.
- del Arco, A.; Morcillo, J.; Martínez-Morales, J.R.; Galián, C.; Martos, V.; Bovolenta, P.; Satrústegui, J. Expression of the aspartate/glutamate mitochondrial carriers aralar1 and citrin during development and in adult rat tissues. Eur. J. Biochem. 2002, 269, 3313–3320.
- Ying, W.; Alano, C.C.; Garnier, P.; Swanson, R.A. NAD+ as a metabolic link between DNA damage and cell death. J. Neurosci. Res. 2005, 79, 216–223.
- Alano, C.C.; Tran, A.; Tao, R.; Ying, W.; Karliner, J.S.; Swanson, R.A. Differences among cell types in NAD(+) compartmentalization: A comparison of neurons, astrocytes, and cardiac myocytes. J. Neurosci. Res. 2007, 85, 3378–3385.
- Pittelli, M.; Formentini, L.; Faraco, G.; Lapucci, A.; Rapizzi, E.; Cialdai, F.; Romano, G.; Moneti, G.; Moroni, F.; Chiarugi, A. Inhibition of nicotinamide phosphoribosyltransferase: Cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J. Biol. Chem. 2010, 285, 34106–34114.
- Agudelo, L.Z.; Ferreira, D.M.S.; Dadvar, S.; Cervenka, I.; Ketscher, L.; Izadi, M.; Zhengye, L.; Furrer, R.; Handschin, C.; Venckunas, T.; et al. Skeletal muscle PGC-1α1 reroutes kynurenine metabolism to increase energy efficiency and fatigue-resistance. Nat. Commun. 2019, 10, 2767.
- Alkan, H.F.; Walter, K.E.; Luengo, A.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Lau, A.N.; Al-Zoughbi, W.; Lewis, C.A.; Thomas, C.J.; Hoefler, G.; et al. Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab. 2018, 28, 706–720.
- Broeks, M.H.; Shamseldin, H.E.; Alhashem, A.; Hashem, M.; Abdulwahab, F.; Alshedi, T.; Alobaid, I.; Zwartkruis, F.; Westland, D.; Fuchs, S.; et al. MDH1 deficiency is a metabolic disorder of the malate-aspartate shuttle associated with early onset severe encephalopathy. Hum. Genet. 2019, 138, 1247–1257.
- Pillarisetti, S. A review of Sirt1 and Sirt1 modulators in cardiovascular and metabolic diseases. Recent Pat. Cardiovasc. Drug Discov. 2008, 3, 156–164.
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580.
- White, A.T.; Schenk, S. NAD(+)/NADH and skeletal muscle mitochondrial adaptations to exercise. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E308–E321.
- Li, H.; Xu, M.; Lee, J.; He, C.; Xie, Z. Leucine supplementation increases SIRT1 expression and prevents mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1234–E1244.
- Phielix, E.; Meex, R.; Moonen-Kornips, E.; Hesselink, M.K.; Schrauwen, P. Exercise training increases mitochondrial content and ex vivo mitochondrial function similarly in patients with type 2 diabetes and in control individuals. Diabetologia 2010, 53, 1714–1721.
- Trevellin, E.; Scorzeto, M.; Olivieri, M.; Granzotto, M.; Valerio, A.; Tedesco, L.; Fabris, R.; Serra, R.; Quarta, M.; Reggiani, C.; et al. Exercise Training Induces Mitochondrial Biogenesis and Glucose Uptake in Subcutaneous Adipose Tissue Through eNOS-Dependent Mechanisms. Diabetes 2014, 63, 2800.
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118.
- Moreno-Santos, I.; Pérez-Belmonte, L.M.; Macías-González, M.; Mataró, M.J.; Castellano, D.; López-Garrido, M.; Porras-Martín, C.; Sánchez-Fernández, P.L.; Gómez-Doblas, J.J.; Cardona, F.; et al. Type 2 diabetes is associated with decreased PGC1α expression in epicardial adipose tissue of patients with coronary artery disease. J. Transl. Med. 2016, 14, 243.
- Yang, S.J.; Choi, J.M.; Kim, L.; Park, S.E.; Rhee, E.J.; Lee, W.Y.; Oh, K.W.; Park, S.W.; Park, C.Y. Nicotinamide improves glucose metabolism and affects the hepatic NAD-sirtuin pathway in a rodent model of obesity and type 2 diabetes. J. Nutr. Biochem. 2014, 25, 66–72.
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007, 26, 1913–1923.
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359.
- van de Weijer, T.; Phielix, E.; Bilet, L.; Williams, E.G.; Ropelle, E.R.; Bierwagen, A.; Livingstone, R.; Nowotny, P.; Sparks, L.M.; Paglialunga, S.; et al. Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans. Diabetes 2015, 64, 1193–1201.
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847.
- Xiang, D.; Zhang, Q.; Wang, Y.T. Effectiveness of niacin supplementation for patients with type 2 diabetes: A meta-analysis of randomized controlled trials. Medicine (Baltimore) 2020, 99, e21235.
- Koh, J.H.; Hancock, C.R.; Han, D.H.; Holloszy, J.O.; Nair, K.S.; Dasari, S. AMPK and PPARβ positive feedback loop regulates endurance exercise training-mediated GLUT4 expression in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E931–E939.
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060.
- Costford, S.R.; Bajpeyi, S.; Pasarica, M.; Albarado, D.C.; Thomas, S.C.; Xie, H.; Church, T.S.; Jubrias, S.A.; Conley, K.E.; Smith, S.R. Skeletal muscle NAMPT is induced by exercise in humans. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E117–E126.
- Costford, S.R.; Brouwers, B.; Hopf, M.E.; Sparks, L.M.; Dispagna, M.; Gomes, A.P.; Cornnell, H.H.; Petucci, C.; Phelan, P.; Xie, H.; et al. Skeletal muscle overexpression of nicotinamide phosphoribosyl transferase in mice coupled with voluntary exercise augments exercise endurance. Mol. Metab. 2018, 7, 1–11.
More
Information
Subjects:
Endocrinology & Metabolism
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
09 Jul 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No