 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dorota Wesół-Kucharska | + 1496 word(s) | 1496 | 2021-07-02 08:58:49 | | | |
| 2 | Amina Yu | + 1 word(s) | 1497 | 2021-07-09 03:12:36 | | | | |
| 3 | Dean Liu | Meta information modification | 1497 | 2021-09-28 06:04:13 | | |
Video Upload Options
Mitochondrial diseases are a heterogeneous group of diseases resulting from energy deficit and reduced adenosine triphosphate (ATP) production due to impaired oxidative phosphorylation. The manifestation of mitochondrial disease is usually multi-organ. Epilepsy is one of the most common manifestations of diseases resulting from mitochondrial dysfunction, especially in children.
1. Introduction
Mitochondria are organelles that are present in almost all cells of the body, which are primarily responsible for producing energy (in the form of ATP) by the process of oxidative phosphorylation (OXPHOS), play a role in the homeostasis of calcium ions, take part in signal transduction between cells by producing reactive oxygen species, and also participate in cell apoptosis [1][2]. Mitochondrial diseases (MDs) represent a clinically and genetically heterogeneous group of diseases with a summary incidence estimated at 1.6:5000 live births, making them the most common diseases among inherited metabolic diseases [2][3]. The clinical picture of MD is very varied, but typically the highly energetic tissues are affected, including the central and peripheral nervous system, skeletal muscles, sense organs, heart, liver, gastrointestinal tract, or endocrine system [2][4][5]. Central nervous system symptoms—regression in development, delayed psychomotor development, or epilepsy—are dominant features in children with MD.
2. Pathophysiology of Epilepsy in Mitochondrial Diseases
Epileptic seizure is a sudden and excessive neural discharge resulting from uncontrolled depolarisation of the neural membrane, and its spread is caused by impaired mechanisms regulating this transmission, e.g., balance between inhibitory (γ-aminobutyric acid, GABA) and stimulatory (glutamic and aspartic acid) neurotransmitters. Maintaining membrane polarisation requires high energy input and primarily involves calcium and sodium channels [6]. The pathophysiology of epilepsy in MD is not fully known (Figure 1).
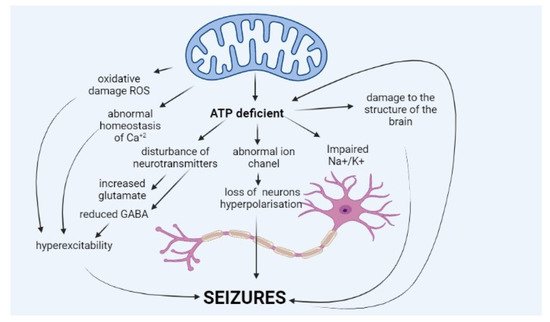
The occurrence of seizures is explained mainly by a deficit of energy, ATP, resulting from impaired oxidative phosphorylation, which is the core of MD. ATP molecules are of key importance for the function of sodium–potassium ATPase (Na+/K+ ATPase), which ensures normal polarisation of the neural membrane and maintenance of resting membrane potential. Impairment in the potential leads to neuronal hyperexcitability, which translates into seizures.
Lack of ATP molecules also results in the reduced potential of GABA-ergic inhibitory neurons, which leads to an impaired balance between excitatory and inhibitory neurons and excessive cell excitation [7][8]. Moreover, ATP deficit leads to increased glutamate release (excitatory neurotransmitter) from astrocytes to synaptic space and disturbance of the glutamate–aspartate transporter [1][7][9]. Other hypotheses explain seizures in MD by abnormal haemostasis of calcium ions, abnormal function of ion channels in the neural membrane, or neurotransmitter disorders [9]. A growing amount of data suggest that seizures occurring in MD patients may be caused by excessive amounts of reactive oxygen species (ROS), resulting from abnormal mitochondrial function [1][10][11].
Moreover, the seizures themselves, especially those that are repeated or long-lasting, lead to mitochondrial dysfunction and escalate energy deficit (epileptic seizures use large amounts of energy), which provokes subsequent seizures and makes them difficult to suppress. Such a self-perpetuating cycle may lead to the development of stroke-resistant episodes common in MD, especially in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) and POLG-related diseases (diseases resulting from damage to DNA polymerase gamma), and the occurrence of such episodes is often related to status epilepticus [12][9].
The degree of heteroplasmy explains only to some extent the severity of mitochondrial diseases. Moreover, as supported by Tranah et al., the accumulation of a rare genetic disease mutation, e.g., m.3243A>G, manifests as several ageing outcomes, and some diseases of ageing may be attributed to the accumulation of mtDNA damage, leading to differing phenotypes [13]. Finally, the role of other genetic variants or polymorphism has to be defined. Still, the provided results showed good evidence for the presence of nuclear genetic factors influencing clinical outcomes in m.3234A>G-related disease [14].
3. Pharmacological Treatment of Epilepsy in Patients with Mitochondrial Disease
Treatment of mitochondrial disorders is a challenge for physicians and researchers. Most interventions and guidelines are related to symptomatic treatment, with supplementation of cofactors, vitamins, or antioxidants, and mild exercises are recommended. Despite numerous studies, the efficacy of most of these interventions has not been confirmed [15][16][17][18][19].
There is also no established scheme of epilepsy treatment in mitochondrial diseases; therefore, general principles of epilepsy treatment are applied [20]. Zonisamide (ZNS) is also safe, but there are few literature reports of patients treated with this medication [21][22][23]. Lamotrigine (LTG) may promote myoclonic seizures and has not always been effective in patients with MD. In most patients, seizures are intractable, which often require polytherapy with two or three medications.
Treatment of status epilepticus is also challenging. The management of MD patients is similar to the management of status epilepticus in other patients, except for avoiding the administration of valproic acid. One must bear in mind that MD patients are more susceptible to the development of propofol infusion syndrome. Although propofol is not contraindicated in patients with MD, caution is recommended while using this drug in this group of subjects [9][24][25].
Regarding contraindicated substances, the only absolutely contraindicated drug in treating epilepsy in MD patients is valproic acid (VPA), which is especially relevant for diseases associated withPOLGpathogenic variants. Experts suggest VPA in MD patients without a pathogenic variant inPOLG, and without liver disease, for the treatment of refractory epilepsy [24]. In patients with mitochondrial depletion syndrome, one should avoid the use of vigabatrin (VGB), which inhibits the conversion of deoxyribonucleoside diphosphate (ADP) to deoxyribonucleoside triphosphate (ATP), whereby it increases mtDNA A list of safe antiepileptic drugs which may be used in patients with mitochondrial disease is presented in Table 2.
| Mitochondria-Safe AEDs | AEDs to Use Carefully | AEDs Which Could Aggravate Myoclonus |
|---|---|---|
| Benzodiazepine [24][26] Gabapentin [24][26] Lacosamide [24][26] Lamotrigine [24][26] Levetiracetam [27][24][26] Oxcarbazepine [27][24][26] Peranpanel [28][24][26] Rufinamide [24][26] Stiripentol [27][24][26] Zonisamide [24][26] |
Valproic acid—contraindicated in POLG mutations [29][15][26] Vigabatrin—may need to be avoided in patients with mtDNA depletion syndromes [15] Topiramate—may worsen acidosis [15] Phenytoin * [30] Carbamazepine * [30] Phenobarbital * [30] |
Valproic acid [22] Phenobarbital [22] Lamotrigine [22] Phenytoin [22] Carbamazepine [22] Oxcarbazepine [22] Vigabatrin [22] Tiagabine [22] Gabapentin [22] Pregabalin [22] |
* Toxic effect on mitochondria outweighs the beneficial effect.
In patients with the m.3243A>G variant, the administration of L-arginine has been confirmed to reduce the incidence of stroke-like episodes and thus reduce the risk of epilepsy and status epilepticus in the course of such episodes [31]. There are also single reports in subjects with epilepsy and Kearns–Sayre syndrome diagnosed with folate (5-methyltetrahydrofolate) deficit, where folic acid supplementation in these subjects was related to improvement in seizure control [32]. Next, high doses of co-enzyme Q10 in patients with primary co-enzyme Q10 deficiency may reduce epileptic seizures in this group of subjects [22].
MD therapy must not exclude such diseases, where it is possible to apply causal treatment or at least mitigation of disease symptoms. Examples of such conditions with specific procedures are presented in Table 3.
| Disease (Gene) |
Clinical Features | Treatment |
|---|---|---|
| Primary co-enzyme Q10 deficiency (COQ2, COQ4, COQ5, COQ6, COQ7, COQ9, PDSS1, PDSS2) | Multisystem involvement with progressive neurological dysfunction, seizures, encephalopathy, stroke-like episodes, cerebellar ataxia, pyramidal dysfunction, cognitive impairment renal failure, and steroid-resistant nephrotic syndrome | High-dose oral CoQ10 supplementation (ranging from 5 to 50 mg/kg/day) [33] |
| Pyruvate dehydrogenase complex (PDHc) deficiency (PDHA1, PDHB, LIAS, PDP1, PDHX, DLAT) |
Epilepsy, developmental delay, ataxia, hypotonia, hypertonia, abnormal eye movements, dystonia, ataxia, axonal neuropathy, and poor feeding | Ketogenic diet 3:1–4:1 and thiamine (50 mg/kg/day, max 300–900 mg/day) [34][35] |
| ACAD9 deficiency (ACAD9) |
Hypertrophic cardiomyopathy, lactic acidosis, exercise intolerance, and occasional seizures | Riboflavin (vitamin B2) 20 mg/kg/day–max 400 mg/day [36] |
| Impairment of thiamine transport and metabolism (SLC19A3, SLC19A2, SLC25A19, TPK1) | Biotin–thiamine-responsive basal ganglia disease or Leigh syndrome; subacute encephalopathy with confusion, dysphagia, dysarthria, seizures, external ophthalmoplegia, and generalised stiffness following a history of febrile illness; progresses to severe quadriparesis, rigidity, dystonia, coma, and death if early treatment is not administered | Biotin (5–10 mg/kg/day) and thiamine (10–40 mg/kg/day, between 300 and 900 mg/day) [37] |
| AGC1 deficiency (aspartate–glutamate carrier isoform 1) (SLC25A12) | Severe hypotonia, arrested psychomotor development, and seizures from a few months of age, a global lack of myelination in the cerebral hemispheres | Ketogenic diet 3:1–4:1 [38] |
| Ethylmalonic encephalopathy (ETHE1) | Early onset, progressive disorder, developmental delay, generalised infantile hypotonia that evolves into hypertonia, spasticity and dystonia; generalised tonic–clonic seizures; and generalised microvascular damage | N-acetylcysteine in combination with metronidazole [39] |
| Beta-hydroxyisobutyryl-CoA deacylase deficiency (HIBCHD) | Progressive neurodegenerative disorder, associated with basal ganglia changes on brain magnetic resonance imaging; elevated hydroxy-C4-carnitine levels | Low-valine and high-carbohydrate diets, antioxidants (co-enzyme Q10, vitamin E, vitamin C), carnitine, and N-acetylcysteine [40] |
4. Prognosis in Epilepsy in Patients with Mitochondrial Disease
Persistent seizures and status epilepticus lead to neural damage, astrocyte gliosis, damage to myelin, and, as a result, brain atrophy [5]. As confirmed by neuroimaging in subjects with MD and epilepsy, this group more often showed brain atrophy than those with MD and without epilepsy [41][42]. The occurrence of seizures in children is related to a worse prognosis regarding development and survival, especially if epilepsy occured at < 1 year. Seizures are often intractable, and epilepsy leads to progressive neurodegenerative changes and epileptic encephalopathy [43][44][45].
References
- Rahman, S. Pathophysiology of mitochondrial disease causing epilepsy and status epilepticus. Epilepsy Behav. 2015, 49, 71–75.
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784.
- Tan, J.; Wagner, M.; Stenton, S.L.; Strom, T.M.; Wortmann, S.B.; Prokisch, H.; Meitinger, T.; Oexle, K.; Klopstock, T. Lifetime risk of autosomal recessive mitochondrial disorders calculated from genetic databases. EBioMedicine 2020, 54, 102730.
- Davison, J.E.; Rahman, S. Recognition, investigation and management of mitochondrial disease. Arch. Dis. Child. 2017, 102, 1082–1090.
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250.
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530.
- Fei, Y.; Shi, R.; Song, Z.; Wu, J. Metabolic Control of Epilepsy: A Promising Therapeutic Target for Epilepsy. Front. Neurol. 2020, 11, 592514.
- Lax, N.Z.; Grady, J.; Laude, A.; Chan, F.; Hepplewhite, P.D.; Gorman, G.; Whittaker, R.G.; Ng, Y.; Cunningham, M.O.; Turnbull, D.M. Extensive respiratory chain defects in inhibitory interneurones in patients with mitochondrial disease. Neuropathol. Appl. Neurobiol. 2016, 42, 180–193.
- Bindoff, L.A.; Engelsen, B.A. Mitochondrial diseases and epilepsy. Epilepsia 2012, 53 (Suppl. S4), 92–97.
- Sanganahalli, B.G.; Herman, P.; Hyder, F.; Kannurpatti, S.S. Mitochondrial calcium uptake capacity modulates neocortical excitability. J. Cereb. Blood Flow Metab. 2013, 33, 1115–1126.
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262.
- Matricardi, S.; Canafoglia, L.; Ardissone, A.; Moroni, I.; Ragona, F.; Ghezzi, D.; Lamantea, E.; Nardocci, N.; Franceschetti, S.; Granata, T. Epileptic phenotypes in children with early-onset mitochondrial diseases. Acta Neurol. Scand. 2019, 140, 184–193.
- Tranah, G.J.; Katzman, S.M.; Lauterjung, K.; Yaffe, K.; Manini, T.M.; Kritchevsky, S.; Newman, A.B.; Harris, T.B.; Cummings, S.R. Mitochondrial DNA m.3243A>G heteroplasmy affects multiple aging phenotypes and risk of mortality. Sci. Rep. 2018, 8, 11887.
- Pickett, S.J.; Grady, J.P.; Ng, Y.S.; Gorman, G.S.; Schaefer, A.M.; Wilson, I.J.; Cordell, H.J.; Turnbull, D.M.; Taylor, R.W.; McFarland, R. Phenotypic heterogeneity in m.3243A>G mitochondrial disease: The role of nuclear factors. Ann. Clin. Transl. Neurol. 2018, 5, 333–345.
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2017, 19.
- Garone, C.; Viscom, C. Towards a therapy for mitochondrial disease: An update. Biochem. Soc. Trans. 2018, 46, 1247–1261.
- Almannai, M.; El-Hattab, A.W.; Ali, M.; Soler-Alfonso, C.; Scaglia, F. Clinical trials in mitochondrial disorders, an update. Mol. Genet. Metab. 2020, 131, 1–13.
- Tinker, R.; Lim, A.Z.; Stefanetti, R.; McFarland, R. Current and Emerging Clinical Treatment in Mitochondrial Disease. Mol. Diagn. Ther. 2021, 25, 181–206.
- Mitochondrial Disease Care Recommendations. Physiotherapy Guidance for People with Mitochondrial Disease. Available online: (accessed on 26 June 2019).
- Rahman, S. Advances in the treatment of mitochondrial epilepsies. Epilepsy Behav. 2019, 101 Pt B, 106546.
- Lamperti, C.; Zeviani, M. Myoclonus epilepsy in mitochondria disorders. Epileptic Disord. 2016, 18, 94–102.
- Finsterer, J. Pharmacotherapeutic management of epilepsy in MERRF syndrome. Expert Opin. Drug Metab. Toxicol. 2019, 20, 1289–1297.
- Su, L.J.; Wang, Y.L.; Han, T.; Qiao, S.; Zang, K.J.; Wu, H.K.; Su, Y.X.; Liu, L.L.; Liu, X.W. Antimyoclonic effect of levetiracetam and clonazepam combined treatment on myoclonic epilepsy with ragged-red fiber syndrome with m.8344A>G Mutation. Chin. Med. J. 2018, 131, 2433–2438.
- De Vries, M.C.; Brown, D.A.; Allen, M.E.; Bindoff, L.; Gorman, G.S.; Karaa, A.; Keshavan, N.; Lamperti, C.; McFarland, R.; Ng, Y.S.; et al. Safety of drug use in patients with a primary mitochondrial disease: An international Delphi-based consensus. J. Inherit. Metab. Dis. 2020, 43, 800–818.
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohent, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701.
- Gruosso, F.; Montano, V.; Simoncini, C.; Siciliano, G.; Mancuso, M. Therapeutical Management and Drug Safety in Mitochondrial Diseases-Update 2020. J. Clin. Med. 2020, 10, 94.
- Ticci, C.; Sicca, F.; Ardissone, A.; Bertini, E.; Carelli, V.; Diodato, D.; Di Vito, L.; Filosto, M.; La Morgia, C.; Lamperti, C.; et al. Mitochondrial epilepsy: A cross-sectional nationwide Italian survey. Neurogenetics 2020, 21, 87–96.
- Santamarina, E.; Alpuente, A.; Maisterra, O.; Sueiras, M.; Sarria, S.; Guzman, L.; Abraira, L.; Salas-Puig, J.; Toledo, M. Perampanel: A therapeutic alternative in refractory status epilepticus associated with MELAS syndrome. Epilepsy Behav. Case Rep. 2019, 11, 92–95.
- Hikmat, O.; Naess, K.; Engvall, M.; Klingenberg, C.; Rasmussen, M.; Tallaksen, C.M.; Brodtkorb, E.; Ostergaard, E.; de Coo, I.F.M.; Pias-Peleteiro, L.; et al. Simplifying the clinical classification of polymerase gamma (POLG) disease based on age of onset; studies using a cohort of 155 cases. J. Inherit. Metab. Dis. 2020, 43, 726–736.
- Finsterer, J. Toxicity of Antiepileptic Drugs to Mitochondria. Handb. Exp. Pharmacol. 2017, 240, 473–488.
- Koga, Y.; Nishioka, Y.; Nishioka, J.; Yatsuga, S.; Povalko, N.; Katayama, K.; Matsuishi, T. MELAS and L-arginine therapy. Mitochondrion 2007, 7, 133–139.
- Pineda, M.; Ormazabal, A.; Lopez-Gallardo, E.; Nascimento, A.; Solano, A.; Herrero, M.D.; Vilaseca, M.A.; Briones, P.; Ibáñez, L.; Montoya, J.; et al. Cerebral folate deficiency and leukoencephalopathy caused by a mitochondrial DNA deletion. Ann. Neurol. 2006, 59, 394–398.
- Salviati, L.; Trevisson, E.; Doimo, M.; Navas, P. Primary Coenzyme Q10 Deficiency. In GeneReviews® [Internet], 1993–2020; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017.
- Bhandary, S.; Aguan, K. Pyruvate dehydrogenase complex deficiency and its relationship with epilepsy frequency-An overview. Epilepsy Res. 2015, 116, 40–52.
- Patel, K.P.; O’Brien, T.W.; Subramony, S.H.; Shuster, J.; Stacpoole, P.W. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol. Genet. Metab. 2012, 105, 34–43.
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rötig, A.; Ardissone, A.; Lombès, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: Is riboflavin supplementation effective? Orphanet J. Rare Dis. 2018, 13, 120.
- Marcé-Grau, A.; Martí-Sánchez, L.; Baide-Mairena, H.; rtigoza-Escobar, J.D.; Pérez-Dueñas, B. Genetic defects of thiamine transport and metabolism: A review of clinical phenotypes, genetics, and functional studies. J. Inherit. Metab. Dis. 2019, 42, 581–597.
- Dahlin, M.; Martin, D.A.; Hedlund, Z.; Jonsson, M.; von Döbeln, U.; Wedell, A. The ketogenic diet compensates for AGC1 deficiency and improves myelination. Epilepsia 2015, 56, e176–e181.
- Viscomi, C.; Burlina, A.B.; Dweikat, I.; Savoiardo, M.; Lamperti, C.; Hildebrandt, T.; Tiranti, V.; Zeviani, M. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat. Med. 2010, 16, 869–871.
- Yamada, K.; Naiki, M.; Hoshino, S.; Kitaura, Y.; Kondo, Y.; Nomura, N.; Kimura, R.; Fukushi, D.; Yamada, Y.; Shimozawa, N.; et al. Clinical and biochemical characterization of 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) deficiency that causes Leigh-like disease and ketoacidosis. Mol. Genet. Metab. Rep. 2014, 16, 455–460.
- Lee, S.; Na, J.H.; Lee, Y.M. Epilepsy in Leigh Syndrome with Mitochondrial DNA Mutations. Front. Neurol. 2019, 10, 496.
- Lee, H.F.; Chi, C.S.; Tsai, C.R.; Chen, C.H. Epileptic seizures in infants and children with mitochondrial diseases. Pediatr. Neurol. 2011, 45, 169–174.
- Lim, A.; Thomas, R.H. The mitochondrial epilepsies. Eur. J. Paediatr. Neurol. 2020, 24, 47–52.
- Khurana, D.S.; Salganicoff, L.; Melvin, J.J.; Hobdell, I.; Valencia, H.H.; Hardison, H.G.; Marks, W.D.; Grover, A.; Legido, A. Epilepsy and respiratory chain defects in children with mitochondrial encephalopathies. Neuropediatrics 2008, 39, 8–13.
- Lee, S.; Baek, M.S.; Lee, Y.M. Lennox-Gastaut Syndrome in Mitochondrial Disease. Yonsei. Med. J. 2019, 60, 106–114.


