 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Weria Pezeshkian | + 2706 word(s) | 2706 | 2021-07-08 04:22:31 | | | |
| 2 | Bruce Ren | -21 word(s) | 2685 | 2021-07-09 03:01:44 | | |
Video Upload Options
Many bacteria secrete toxic protein complexes that modify and disrupt essential processes in the infected cell that can lead to cell death. To conduct their action, these toxins often need to cross the cell membrane and reach a specific substrate inside the cell. The investigation of these protein complexes is essential not only for understanding their biological functions but also for the rational design of targeted drug delivery vehicles that must navigate across the cell membrane to deliver their therapeutic payload. Despite the immense advances in experimental techniques, the investigations of the toxin entry mechanism have remained challenging. Computer simulations are robust complementary tools that allow for the exploration of biological processes in exceptional detail.
1. Introduction
2. Computational Methods
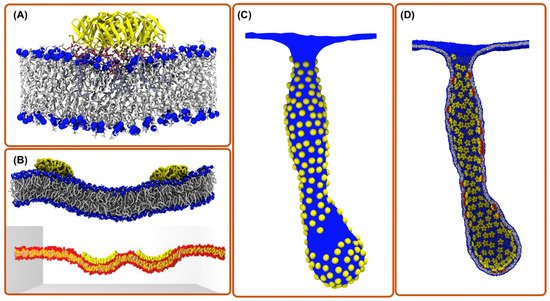
3. All-Atom Molecular Dynamics Simulations
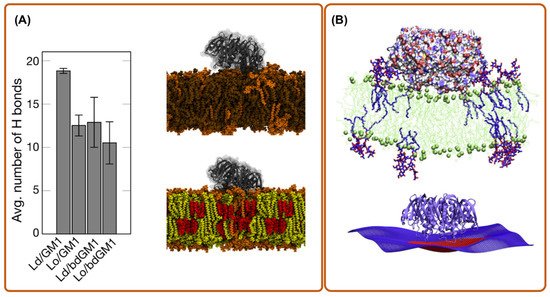
4. Coarse-Grain Simulations
5. Summary
Computer modeling techniques have become an indispensable investigatory tool for exploring biological processes on a wide range of time and length scales. Owing to the rapid technological progress in hardware and software, and efficient algorithms and force fields, they have emerged as a new field, filling the gap between the theoretical and experimental investigations. Indeed, the mechanism of action of bacterial toxins is a good example. In particular, with the development of new multiscale simulation approaches, valuable information about their mechanism of action and entry pathway has been obtained. Multiscale simulations provide a scheme for analyzing the cascade of successive complex processes of toxin action from the molecular details to the effect of membrane structural deformations and fluctuations. With the fast progress in the multiscale simulation techniques, these methods will become a standard tool complementary to experiments in the research of toxins. Additionally, simulations can provide certain collective properties emerging from molecular features that can be used as tuning parameters in the rational design and engineering of drug delivery vehicles.References
- Schiavo, G.; Van Der Goot, F.G. The bacterial toxin toolkit. Nat. Rev. Mol. Cell Biol. 2001, 2, 530–537.
- Klenow, M.B.; Jeppesen, J.C.; Simonsen, A.C. Membrane rolling induced by bacterial toxins. Soft Matter 2020, 16, 1614–1626.
- Piper, S.J.; Brillault, L.; Rothnagel, R.; I Croll, T.; Box, J.K.; Chassagnon, I.; Scherer, S.; Goldie, K.N.; A Jones, S.; Schepers, F.; et al. Cryo-EM structures of the pore-forming A subunit from the Yersinia entomophaga ABC toxin. Nat. Commun. 2019, 10, 1–12.
- Bacia, K.; Scherfeld, D.; Kahya, N.; Schwille, P. Fluorescence Correlation Spectroscopy Relates Rafts in Model and Native Membranes. Biophys. J. 2004, 87, 1034–1043.
- Römer, W.; Berland, L.; Chambon, V.; Gaus, K.; Windschiegl, B.; Tenza, D.; Aly, M.R.E.; Fraisier, V.; Florent, J.-C.; Perrais, D.; et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 2007, 450, 670–675.
- Chang, K.J.; Benett, V.; Cuatrecasas, P. Membrane receptors as general markers for plasma membrane isolation procedures. The use of 125-I-labeled wheat germ agglutinin, insulin, and cholera toxin. J. Biol. Chem. 1975, 250, 488–500.
- Shogomori, H.; Futerman, A. Cholera Toxin Is Found in Detergent-insoluble Rafts/Domains at the Cell Surface of Hippocampal Neurons but Is Internalized via a Raft-independent Mechanism. J. Biol. Chem. 2001, 276, 9182–9188.
- Arumugam, S.; Schmieder, S.; Pezeshkian, W.; Becken, U.; Wunder, C.; Chinnapen, D.; Ipsen, J.H.; Kenworthy, A.K.; Lencer, W.; Mayor, S.; et al. Ceramide structure dictates glycosphingolipid nanodomain assembly and function. Nat. Commun. 2021, 12, 3675–3687.
- Hammond, A.T.; Heberle, F.; Baumgart, T.; Holowka, D.; Baird, B.; Feigenson, G.W. Crosslinking a lipid raft component triggers liquid ordered-liquid disordered phase separation in model plasma membranes. Proc. Natl. Acad. Sci. USA 2005, 102, 6320–6325.
- Johannes, L.; Wunder, C.; Shafaq-Zadah, M. Glycolipids and Lectins in Endocytic Uptake Processes. J. Mol. Biol. 2016, 428, 4792–4818.
- Johannes, L. Shiga Toxin—A Model for Glycolipid-Dependent and Lectin-Driven Endocytosis. Toxins 2017, 9, 340.
- Doosti, B.A.; Pezeshkian, W.; Bruhn, D.S.; Ipsen, J.; Khandelia, H.; Jeffries, G.D.M.; Lobovkina, T. Membrane Tubulation in Lipid Vesicles Triggered by the Local Application of Calcium Ions. Langmuir 2017, 33, 11010–11017.
- Pezeshkian, W.; Marrink, S.J. Simulating realistic membrane shapes. Curr. Opin. Cell Biol. 2021, 71, 103–111.
- Enkavi, G.; Javanainen, M.; Kulig, W.; Róg, T.; Vattulainen, I. Multiscale Simulations of Biological Membranes: The Challenge To Understand Biological Phenomena in a Living Substance. Chem. Rev. 2019, 119, 5607–5774.
- Joshi, H.; Bhatia, D.; Krishnan, Y.; Maiti, P.K. Probing the structure and in silico stability of cargo loaded DNA icosahedra using MD simulations. Nanoscale 2017, 9, 4467–4477.
- Patmanidis, I.; De Vries, A.H.; Wassenaar, T.A.; Wang, W.; Portale, G.; Marrink, S.J. Structural characterization of supramolecular hollow nanotubes with atomistic simulations and SAXS. Phys. Chem. Chem. Phys. 2020, 22, 21083–21093.
- Khan, F.I.; Wei, D.; Gu, K.-R.; Hassan, I.; Tabrez, S. Current updates on computer aided protein modeling and designing. Int. J. Biol. Macromol. 2016, 85, 48–62.
- Souza, P.C.T.; Thallmair, S.; Conflitti, P.; Ramírez-Palacios, C.; Alessandri, R.; Raniolo, S.; Limongelli, V.; Marrink, S.J. Protein–ligand binding with the coarse-grained Martini model. Nat. Commun. 2020, 11, 1–11.
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sønder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623–1634.
- Florentsen, C.D.; Kamp-Sonne, A.; Moreno-Pescador, G.; Pezeshkian, W.; Zanjani, A.A.H.; Khandelia, H.; Nylandsted, J.; Bendix, P.M. Annexin A4 trimers are recruited by high membrane curvatures in giant plasma membrane vesicles. Soft Matter 2021, 17, 308–318.
- Pezeshkian, W.; Hansen, A.G.; Johannes, L.; Khandelia, H.; Shillcock, J.; Kumar, P.B.S.; Ipsen, J.H. Membrane Invagination Induced by Shiga toxin B-subunit: From Molecular Structure to Tube Formation. Soft Matter 2016, 12, 5164–5171.
- Pezeshkian, W.; Gao, H.; Arumugam, S.; Becken, U.; Bassereau, P.; Florent, J.-C.; Ipsen, J.H.; Johannes, L.; Shillcock, J.C. Mechanism of Shiga Toxin Clustering on Membranes. ACS Nano 2017, 11, 314–324.
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73.
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130.
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25.
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174.
- Pezeshkian, W.; Nåbo, L.J.; Ipsen, J.H. Cholera toxin B subunit induces local curvature on lipid bilayers. FEBS Open Bio 2017, 7, 1638–1645.
- Basu, I.; Mukhopadhyay, C. Insights into Binding of Cholera Toxin to GM1 Containing Membrane. Langmuir 2014, 30, 15244–15252.
- Pezeshkian, W.; Chaban, V.V.; Johannes, L.; Shillcock, J.; Ipsen, J.; Khandelia, H. The effects of globotriaosylceramide tail saturation level on bilayer phases. Soft Matter 2015, 11, 1352–1361.
- Flores-Canales, J.C.; Kurnikova, M.; Maria, K. Microsecond Simulations of the Diphtheria Toxin Translocation Domain in Association with Anionic Lipid Bilayers. J. Phys. Chem. B 2015, 119, 12074–12085.
- Ghatak, C.; Rodnin, M.V.; Vargas-Uribe, M.; McCluskey, A.J.; Flores-Canales, J.C.; Kurnikova, M.; Ladokhin, A.S. Role of Acidic Residues in Helices TH8–TH9 in Membrane Interactions of the Diphtheria Toxin T Domain. Toxins 2015, 7, 1303–1323.
- Flores-Canales, J.C.; Simakov, N.A.; Kurnikova, M. Microsecond Molecular Dynamics Simulations of Diphtheria Toxin Translocation T-Domain pH-Dependent Unfolding in Solution. bioRxiv 2019, 572040.
- Ladokhin, A.S. pH-triggered conformational switching along the membrane insertion pathway of the diphtheria toxin T-domain. Toxins 2013, 5, 1362–1380.
- Rissanen, S.; Grzybek, M.; Orłowski, A.; Róg, T.; Cramariuc, O.; Levental, I.; Eggeling, C.; Sezgin, E.; Vattulainen, I. Phase Partitioning of GM1 and Its Bodipy-Labeled Analog Determine Their Different Binding to Cholera Toxin. Front. Physiol. 2017, 8, 252.
- Lingwood, D.; Binnington, B.; Róg, T.; Vattulainen, I.; Grzybek, M.; Ünal, C.; Lingwood, C.A.; Simons, K. Cholesterol modulates glycolipid conformation and receptor activity. Nat. Chem. Biol. 2011, 7, 260–262.
- Manna, M.; Javanainen, M.; Monne, H.M.-S.; Gabius, H.-J.; Rog, T.; Vattulainen, I. Long-chain GM1 gangliosides alter transmembrane domain registration through interdigitation. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 870–878.
- Merritt, E.A.; Kuhn, P.; Sarfaty, S.; Erbe, J.L.; Holmes, R.K.; Hol, W.G.J. The 1.25 Å resolution refinement of the cholera toxin B-pentamer: Evidence of peptide backbone strain at the receptor-binding site. J. Mol. Biol. 1998, 282, 1043–1059.
- Johannes, L.; Parton, R.; Bassereau, P.; Mayor, S. Building endocytic pits without clathrin. Nat. Rev. Mol. Cell Biol. 2015, 16, 311–321.
- Kabbani, A.M.; Raghunathan, K.; Lencer, W.I.; Kenworthy, A.K.; Kelly, C.V. Structured clustering of the glycosphingolipid GM1 is required for membrane curvature induced by cholera toxin. Proc. Natl. Acad. Sci. USA 2020, 117, 14978–14986.
- Groza, R.; Ewers, H. Membrane deformation by the cholera toxin beta subunit requires more than one binding site. Proc. Natl. Acad. Sci. USA 2020, 117, 17467–17469.
- Ewers, H.; Römer, W.; Smith, A.E.; Bacia, K.; Dmitrieff, S.; Chai, W.; Mancini, R.; Kartenbeck, J.; Chambon, V.; Berland, L.; et al. GM1 structure determines SV40-induced membrane invagination and infection. Nat. Cell Biol. 2009, 12, 11–18.
- Ivashenka, A.; Wunder, C.; Chambon, V.; Sandhoff, R.; Jennemann, R.; Dransart, E.; Podsypanina, K.; Lombard, B.; Loew, D.; Lamaze, C.; et al. Glycolipid-dependent and lectin-driven transcytosis in mouse enterocytes. Commun. Biol. 2021, 4, 1–15.
- Lakshminarayan, R.; Wunder, C.; Becken, U.; Howes, M.; Benzing, C.; Arumugam, S.; Sales, S.; Ariotti, N.; Chambon, V.; Lamaze, C.; et al. Galectin-3 drives glycosphingolipid-dependent biogenesis of clathrin-independent carriers. Nat. Cell Biol. 2014, 16, 592–603.
- Renard, H.-F.; Tyckaert, F.; Giudice, C.L.; Hirsch, T.; Valades-Cruz, C.A.; Lemaigre, C.; Shafaq-Zadah, M.; Wunder, C.; Wattiez, R.; Johannes, L.; et al. Endophilin-A3 and Galectin-8 control the clathrin-independent endocytosis of CD166. Nat. Commun. 2020, 11, 1–13.
- Ingólfsson, H.I.; Lopez, C.A.; Uusitalo, J.J.; De Jong, D.H.; Gopal, S.M.; Periole, X.; Marrink, S.J. The power of coarse graining in biomolecular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 225–248.
- Saunders, M.G.; Voth, G.A. Coarse-Graining Methods for Computational Biology. Annu. Rev. Biophys. 2013, 42, 73–93.
- Lipowsky, R.; Brinkmann, M.; Dimova, R.; Haluska, C.; Kierfeld, J.; Shillcock, J. Wetting, budding, and fusion—morphological transitions of soft surfaces. J. Phys. Condens. Matter 2005, 17, S2885–S2902.
- Johannes, L.; Pezeshkian, W.; Ipsen, J.H.; Shillcock, J.C. Clustering on Membranes: Fluctuations and More. Trends Cell Biol. 2018, 28, 405–415.
- Reynwar, B.J.; Illya, G.; Harmandaris, V.A.; Müller, M.M.; Kremer, K.; Deserno, M. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature 2007, 447, 461–464.
- Cooke, I.; Kremer, K.; Deserno, M. Tunable generic model for fluid bilayer membranes. Phys. Rev. E 2005, 72 Pt 1, 011506.
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Badaczewska-Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936.
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388.
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824.
- Liu, Y.; Barnoud, J.; Marrink, S.J. Gangliosides Destabilize Lipid Phase Separation in Multicomponent Membranes. Biophys. J. 2019, 117, 1215–1223.
- Sridhar, A.; Kumar, A.; DasMahapatra, A.K. Multi-scale molecular dynamics study of cholera pentamer binding to a GM1-phospholipid membrane. J. Mol. Graph. Model. 2016, 68, 236–251.
- Kociurzynski, R.; Makshakova, O.N.; Knecht, V.; Römer, W. Multiscale Molecular Dynamics Studies Reveal Different Modes of Receptor Clustering by Gb3-Binding Lectins. J. Chem. Theory Comput. 2021, 17, 2488–2501.
- Flores-Canales, J.C.; Vargas-Uribe, M.; Ladokhin, A.S.; Kurnikova, M. Membrane Association of the Diphtheria Toxin Translocation Domain Studied by Coarse-Grained Simulations and Experiment. J. Membr. Biol. 2015, 248, 529–543.


