 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jagdeep Sandhu | + 4718 word(s) | 4718 | 2021-06-07 08:20:03 | | | |
| 2 | Vivi Li | Meta information modification | 4718 | 2021-07-08 08:38:25 | | |
Video Upload Options
Inflammasomes are molecular platforms that are formed and activated by a host in response to pathogen- and damage-associated molecular patterns as well as cellular injury and stress. Inflammasome activation is benefical to the host as it plays a pivotal role in the clearance of the pathogen and restore tissue homeostasis. However, aberrant activation of the NLRP3 inflammasome by a wide variety of sterile triggers, including misfolded protein aggregates drives pathological sterile inflammation and is associated with several neuromuscular diseases. Assembly of the NLRP3 inflammasome leads to the caspase-1-mediated proteolytic cleavage and secretion of key proinflammatory cytokines, interleukin (IL)-1β and IL-18, and a lytic form of cell death known as pyroptosis. These cytokines further amplify inflammatory responses by activating various signaling cascades, leading to the recruitment of immune cells and overproduction of proinflammatory cytokines and chemokines, resulting in a vicious cycle of chronic inflammation and tissue damage.
1. Introduction
Skeletal muscle is a secretory organ that releases cytokines (also called myokines) which act as signaling messengers between the muscle and other organs in the endocrine system; thus, it plays an important role both in physiological and pathological processes [1]. In fact, at the early stage of muscle regeneration, following muscle damage, infiltrating immune cells (i.e., mast cells and neutrophils) clear the damaged myofibers and secrete cytokines to recruit macrophages, to regulate the inflammatory response. These cytokines are known to induce muscle wasting by stimulating proteolysis, thereby tipping the fulcrum towards protein degradation, leading in turn to impairment of muscle regeneration [2].
The Nod-like receptor (NLRs) proteins represent a family of conserved cytoplasmic pattern recognition receptors that act as immune sensors of the innate immune system. NLRs are multiprotein complexes capable of activating an inflammatory response; amongst them, NLRP1, NLRP3, NLRP6, NLRP7, NLRP12, NLRC4 (NLR family CARD domain-containing protein 4) and NAIP (neuronal apoptosis inhibitory protein) accomplish this via the formation of inflammasomes [3]. The inflammasome complex consists of three components, namely a sensor molecule, the adaptor ASC and the effector caspase-1. The stimulation and assembly of inflammasomes induces a chain-reaction of caspase 1-dependent proteolytic cleavage, along with the maturation and secretion of proinflammatory cytokines, namely, IL-1β and IL-18, resulting in a highly inflammatory form of programmed cell death, distinct from apoptosis, called pyroptosis. This process leads to plasma membrane rupture and the release of proinflammatory intracellular contents, including inflammasome components, into the extracellular milieu to promote chemotaxis and infiltration of innate immune cells at the sites of tissue damage [4].
2. NLRP3 Inflammasome
The NLRP3 inflammasome is one of the most studied and characterized inflammasomes due to its unique and wide diversity of stimuli, and its suggested involvement in a variety of disorders [5][6][7]. Although the NLRP3 inflammasome is important for detecting microbial and host-derived danger signals and promoting tissue repair, its over-activation may lead to severe impairments (i.e., swelling, tissue damage, internal bleeding and respiratory disablement) [7]. In this review we focus primarily on the involvement of the NLRP3 inflammasome in neuromuscular diseases.
The NLRP3 inflammasome is crucial for initiating innate immune responses [8]. The stimulation, assembly, and proper functioning of the inflammasome are dependent on a variety of key players. Many excellent reviews describe in detail the fundamental steps of NLRP3 inflammasome formation and activation [4][9][10][11]. Therefore, only a brief summary is provided below.
It is also well established that the NACHT domain is critical for NLRP3 function and self-association [12]. The assembly of inflammasome is initiated by recruitment of procaspase-1 and proximity-induced oligomerization and autoactivation, resulting in caspase-1 cleavage and release of active caspase-1. Consequently, caspase-1 cleaves pro-interleukin 1-beta (pro-IL-1β) and pro-interleukin 18 (pro-IL-18) into biologically active mature forms of IL-1β and IL-18. Recent research has shown that pyroptosis is executed through activation and cleavage of caspase-1 and -11 mediated cleavage of gasdermin D (GSDMD). The N-terminal fragments of GSDMD binds to the cell membrane and forms pores, followed by cellular swelling, plasma membrane rupture and release of cellular contents into the extracellular milieu [13][14]. The exact mechanisms involved in the execution of pyroptosis and regulation of host defense responses remain unclear.
Although ASC has been identified as a key adaptor that links the PYD-domain-containing NLR receptors, such as NLRP3, to the pro-caspase-1 via its PYD and CARD domains [15], it is also responsible for the formation of ASC specks (called pyroptosomes) [16]. The PYD–PYD self-association drives dimerization of ASC and permits further assembly of supramolecular insoluble aggregates independently of caspase-1 [16]. On the other hand, the CARD domain of ASC plays an important role in linking individual ASC filaments to create dense specks [17]. In addition, these specks have been shown to amplify and perpetuate inflammasome signaling by creating many caspase-1 activation sites [17].
The NLRP3 inflammasome can be activated in response to a diverse array of pathogenic and environmental stimuli. In fact, members of the NLR family, including NLRP3, PYRIN and PYHIN family proteins (recognition receptors such as absent in melanoma 2 (AIM2), recently reported to play roles in cell cycle, tumor suppression and transcriptional regulation), have all been shown to assemble inflammasomes in response to cytosolic pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). PAMPs include lipopolysaccharides (LPS), flagellin, viral DNA and RNA whereas DAMPs include S100 proteins, heat shock proteins and High mobility group box 1 protein released from dying cells as well as changes in pH, ion fluxes, extracellular ATP, reactive oxygen species (ROS) and uric acid crystals [18]. The exact mechanism by which NLRP3 is activated by a vast array of pathogenic stimuli is unknown, and future research is needed to uncover the mechanisms involved.
Generally, the activation of NLRP3 inflammasome requires two types of signals: an initial priming signal, followed by an activating signal. Among these, phosphorylation, sumoylation, ubiquitination, alkylation, acetylation and nitration have been linked to NLRP3 activation in most cell types [10]. The second signal, being the activation signal, varies significantly and is stimulated by PAMPs and DAMPs such as extracellular ATP via the ligand-gated ion channel belonging to the purinergic type 2 (P2X7) receptor [19][20]. Many signals are triggered by potassium efflux (K+) and other ionic fluxes (i.e., Ca2+ and Cl−), uric acid crystals or particles (asbestos) causing lysosomal disruption; these are signals causing mitochondrial damage resulting in the release of ROS and mitochondrial (mt) DNA, and changes in the trans-Golgi network [9].
Emerging evidence indicates that protein–protein interactions are vital to NLRP3 function. Interestingly, priming signaling by LPS increases NLRP3–NEK7 interactions. This distinct link between NLRP3 and NEK7 is an electrostatic complementarity interaction caused by NLRP3 being overall negatively charged while NEK-7 is positively charged [21]. NLRP3 and NEK7 can form a large oligomeric complex that leads to inflammasome assembly, as NEK7 forms interactions between adjacent NLRP3 subunits.
The NLRP3 inflammasome has also been shown to bind other PYD containing proteins such as pyrin-only proteins (POPs), a family of proteins consisting of single PYD domains. These proteins are small cytoplasmic decoy proteins that regulate the inflammasome by either activating or inhibiting key players of the inflammasome Thus, POPs acts as part of a negative feedback mechanism permitting host defense and early inflammation, but also resolves inflammasome events in the long-term [22]. Like POPs, COPs can both inhibit and activate the inflammasome.
3. NLRP3 Inflammasome in Muscles
The NLRP3 inflammasome is expressed in the muscles of humans and mice. In addition, primary skeletal muscle cells contain TLR receptors (TLR-2, TLR-4) and the P2X7 receptor, two vital players for priming and activating the inflammasome signaling pathway. Skeletal muscle cells also have the ability to secrete IL-1β in response to treatments with LPS and benzylated ATP, suggesting that similar to immune cells, muscle cells can participate in inflammasome formation [23]. NLRP3 inflammasomes have also been shown to be active in myocytes and are upregulated in degenerating muscles, muscle atrophy, muscle loss and myopathies [8][23][24].
A recent study from Liu et al. described the role of NLRP3 in muscle wasting and atrophy via angiotensin II (ang II) mechanistic signaling. Ang II expression levels are increased in patients suffering from chronic kidney disease or heart failure who display symptoms of muscle loss and muscle wasting [25][26]. WT mice treated with Ang II presented a muscle wasting and weight loss phenotype along with decreased muscle performance, which was reverted in the NLRP3-KO mice [25]. Another study has demonstrated that IL-1β increases in muscle can also stimulate the expression of atrophy markers, MuRF1 and atrogin-1, supporting the involvement of inflammasomes in muscle atrophy [27][28][29].
More importantly, mutations in the NLRP3 gene (coding for cryopyrin) are associated with a group of clinically distinct disorders known as cryopyrin-associated periodic syndromes (CAPS) and encompass a spectrum of phenotypes described as familial cold autoinflammatory syndrome, Muckle–Wells syndrome and neonatal-onset multisystem inflammatory disease. These diseases are characterized by systemic inflammation with elevated levels of proinflammatory mediators, fever, blood neutrophilia, and increased neutrophil infiltration in the skin, joints, muscles, and cerebrospinal fluid. As described above, activation of the NLRP3 inflammasome typically requires two signals; however, in CAPS patients, the gain-of-function decreases the activation threshold to only one signal [30]. Increased serum levels of extracellular oligomeric ASC have been found in patients with active CAPS, but not in patients with other inherited autoinflammatory diseases [31].
Due to this convincing evidence connecting the NLRP3 inflammasome to muscle wasting, investigators have spent considerable efforts on determining whether NLRP3 inflammasome signaling is also involved in neuromuscular and neurodegenerative diseases associated with muscular dysfunction. The involvement of NLRP3 in these disorders is discussed below.
Duchenne muscular dystrophy (DMD) is a disorder caused by deletions or mutations in the dystrophin gene, preventing the production of functional dystrophin protein in skeletal muscle fibers, the diaphragm and the heart [32][33][34]. These patients suffer from severe muscle degeneration which results in muscle weakness, respiratory impairment, and cardiomyopathy. Loss of ambulation arises at 12 years of age and progresses until the age of 16–18 years, after which ambulation is lost [35][36]. With multidisciplinary care, including ventilatory support and cardioprotective management, many patients with DMD can live into their fourth decade of life [37].
In muscles, dystrophin binds the dystrophin-associated protein complex (DAPC) at the muscle membrane and the actin cytoskeleton, which provides muscle membrane stability [38]. Activation of NF-κB is known to increase the expression of cell adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule (VCAM-1) and E-selection, leading to leukocyte adhesion and transmigration to the sites of inflammation [39]. Accordingly, atypical increases of immune cells (e.g., neutrophils, T lymphocytes) and inflammatory macrophages arise in the muscle, provoking chronic inflammation, necrosis and replacement of the muscle by connective tissue [1][40][41] (Figure). Other possible therapeutic strategies aimed at targeting signaling pathways to improve functional outcome of dystrophic muscles can be designed to target endogenous genes to restructure the DAPC, and tackle inflammation, calcium imbalance, fibrosis and oxidative stress [1][42][43].
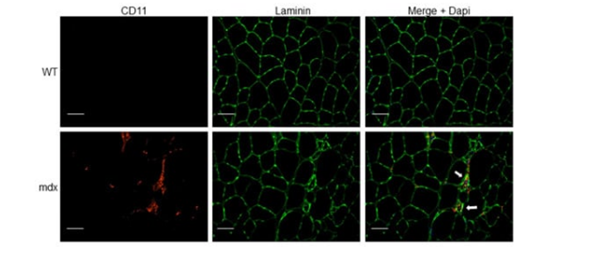
Figure. Immune cell infiltration in dystrophic muscle tissue. Tibialis anterior (TA) muscle cryosections from wild-type (WT) and mdx mice were immunostained with anti-CD11b (myeloid cell marker) and anti-laminin (muscle fiber membrane stain) antibodies. Nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI, blue). Arrows indicate myeloid cell infiltrate (red) associated with focal invasion of muscle fibers (green). Scale bars, 50 μm.
Glucocorticoids have anti-inflammatory effects, in part, by suppressing NF-κB and activator protein-1 (AP-1), which inhibits coactivator molecules responsible for acetylating histones involved in switching on transcription of inflammatory genes [44]. This indicates that alternate anti-inflammatory drugs should be further investigated for DMD. The results from this study showed that the NSAIDs improved inflammation, fiber necrosis and muscle morphology, and aspirin in particular ameliorated resistance to muscle fatigue [45]. Another NSAID, celecoxib (Celebrex), markedly ameliorated muscle fiber integrity, improved muscle function and decreased immune cell infiltration in mdx mice [46].
Although inflammation is a key component of muscle repair, chronic inflammation results in progressive muscle degeneration and weakness. Upstream receptors of inflammasomes, TLRs, including TLR-1, 2, 3, 4, 7, 8 and 9, are also expressed in a variety of skeletal muscles at different levels in mdx mice [47]. Histopathological studies have shown that abnormal fiber size distribution is a hallmark of dystrophic muscles [48][49]. NLRP3 knockdown in mdx mice restores a more equal distribution in muscle fiber size and significantly reduces centrally nucleated myofibers (indicative of fewer regenerating fibers).
In contrast to control mice, transgenic mdx mice overexpressing adiponectin showed decreased inflammation and oxidative stress markers involved in dystrophic muscle damage [50]. Along these lines, a recent publication from Boursereau et al. explored the effects of NLRP3 and adiponectin in muscle, in a DMD context [8]. This study also demonstrated that similarly to what was seen in the transgenic mdx/NLRP3-KO mice, the adiponectin-mediated decrease in NLRP3 promoted beneficial effects on the dystrophic phenotype of mdx mice. These findings reveal the potential involvement of NLRP3 in DMD pathogenesis and adiponectin as a potential therapeutic approach to target inflammation in DMD.
Another potential NLRP3 target characterized in dystrophic mice is ghrelin. This hormone, typically involved in regulating appetite, has been shown to induce anti-inflammatory activity in many inflammatory diseases [51][52][53]. This treatment also improved motor performance as determined by in vivo behavioral testing and morphological assessment of pathology in the mdx muscles. Furthermore, the inhibition of NLRP3 inflammasome by ghrelin was partly mediated by the suppression of janus kinase 2/signal transducer and activator of transcription 3 (JAK2-STAT3) and the p38 mitogen-activated protein kinase signaling pathway [54], typically involved in modulation of inflammation, hematopoiesis, immunity, cell growth, differentiation and proliferation [55].
Although the studies discussed above highlight the importance of NLRP3 in DMD and other dystrophies (described below), contradicting results from a recent study demonstrate that genetic disruption of the inflammasome central adaptor ASC has minimal effects on the phenotype of mdx mice [56]. Therefore, further experimentation is warranted to resolve these discrepancies and delineate the mechanisms involved.
Limb–girdle muscular dystrophy (LGMD) is a genetically and clinically heterogeneous family of rare progressive muscle disorders [57][58]. LGMD disorders are caused by genetic variants resulting in aberrant synthesis of proteins involved in different parts of the muscle fiber, such as the sarcolemma (muscle fiber membrane), sarcoplasm, sarcomere, nucleus and the extracellular matrix [59][60]. Dysferlin is responsible for fusion of repair vesicles with the plasma membrane to repair the damaged muscle membrane, and has been suggested to be involved in the control of muscle inflammation [61][62][63]. Dysferlin expression has also been directly correlated with the ability of mitochondria to improve the repair of the muscle fiber membrane [64].
Mild myofiber damage in dysferlin-deficient muscle is sufficient to activate inflammatory signaling, which may contribute to the progression of the disorder. Indeed, biopsies from LGMD2B patient muscle contained higher levels of protein components of the inflammasome, including NLRP3, ASC and pro-caspase 1, compared to healthy subjects. Treatment of primary skeletal muscle cells from dysferlin-deficient mice with inflammasome activators such as LPS and benzylated ATP led to more secretion of IL-1β into the culture supernatant compared to untreated muscle cells [23]. Thus, with membrane fragility and decreased muscle fiber integrity, as in LGMD2B, the damaged muscle cells are more susceptible to NLRP3 inflammasome activation and the inflammatory response.
The entry of Ca2+ ions due to sarcolemmal tears or the activation of leaky calcium channels by sarcolemmal stretching creates calcium excess in the muscle fibers and in the mitochondria, causing swelling [65][66]. Interestingly, it has been shown that an increase in cytosolic Ca2+ promotes NLRP3 inflammasome activation. It is also suggested that the increase of cytosolic Ca2+ may provoke Ca2+ overloading of mitochondria, resulting in mitochondrial dysfunction that leads to NLRP3 inflammasome activation [67]. Thus, it does not come as a surprise that dystrophic muscles of LGMD2B and DMD patients that have membrane instability and increased Ca2+.
It is unclear whether inflammasome activation seen in dysferlinopathies is only due to sarcolemmal wounding of the muscle fibers or whether it is also caused by lysosomal dysfunction. Dysferlin-containing vesicles have been shown to move along microtubules via kinesin motor proteins, and fuse with lysosomes in response to membrane damage, to create large wound sealing vesicles [68]. In addition, lysosomal destabilization and rupture increases the production of mitochondrial ROS and NLRP3 activation, and promotes the release of IL-1β and IL-18 in both HUVEC and THP-1 monocytes [69][70]. Hence, dysferlin deficiency may result in increased stimulation of NLRP3 inflammasome via lysosomal dysfunction.
4. Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS, also known as motor neuron disease and Lou Gehrig’s disease) is an adult-onset, progressive neurodegenerative disease affecting approximately 2 per 100,000 individuals world-wide, with a higher incidence among men than women. It is characterized by the selective degeneration of motor neurons in the brain, spinal cord and skeletal muscles, leading to severe muscle atrophy and eventually paralysis [71]. The disorder initiates with focal weaknesses but spreads and affects most muscles, leading to paralysis of the whole body. Due to the large number of causative factors that are suggested to increase in risk of developing ALS, disease pathology and clinical presentation of ALS are markedly heterogeneous. The rate of disease progression is very aggressive, and death typically occurs within 2–5 years post-diagnosis, generally due to respiratory failure [72]. The vast majority of ALS cases (>90%) are sporadic, but ~5–10% of the cases are familial [73]. The pathological hallmarks of ALS include the presence of intraneuronal inclusions composed of insoluble aggregated proteins [74].
The molecular mechanisms involved in the initiation and progression of ALS remain unclear; however, mutations in the genes that are important for neuronal survival and function have been identified in patients with the familial and sporadic forms of the disease. A hexanucleotide repeat expansion (GGGGCC) mutation in the non-coding region of the non-coding chromosome 9 open reading frame 72 (C9ORF72) gene is the most common genetic cause of ALS patients and is present in nearly 50% of patients with familial ALS, and 5–10% of patients with sporadic ALS [75]. Although the molecular mechanisms by which the C9ORF72 protein causes neurodegeneration has not been fully elucidated, both loss-of-function through haploinsufficiency and gain-of-function through accumulation of toxic peptides have been implicated. Knockdown of C9ORF72 protein using silencing RNA dysregulated autophagy and inhibited endocytosis, supporting its role in endosomal trafficking and protein degradation [76]. Mutations in the Cu/Zn superoxide dismutase (SOD1) gene are another set of causes of the familial form of the disease; both gain-of-function and loss-of-function mutations have been reported [77][78]. Since the discovery of the first missense mutation in 1993 in the SOD1 gene, advances in human genetics have identified more than 185 SOD1 variants [79]. SOD1 is a ubiquitously expressed enzyme that is found in the cytoplasm and intermembrane space of mitochondria. It is known to bind Cu2+ and Zn2+ ions and convert highly reactive and toxic superoxide radicals to oxygen and hydrogen peroxide, considered to be the less toxic species. Hydrogen peroxide is subsequently converted to water and oxygen in the presence of catalase, thereby promoting an antioxidant defense mechanism in the cell [76]. It has been suggested that mutations in SOD1 result in conformational and functional changes, leading to the inability of the enzyme to scavenge superoxide radicals, leading to oxidative stress. Recent progress in ALS research has shown that an SOD1 mutation contributes to ALS pathology by causing toxic gain-of-function of the mutated protein, in turn leading to misfolding and oligomerization.
Familial ALS has also been linked to mutations in more than 50 separate genes that are known as disease-modifying or causative of ALS, of which TAR DNA-binding protein 43 kDa (TDP-43, encoded by TARDBP) fused in sarcoma (FUS) and C9ORF72, as described above, have been most extensively characterized. A growing body of evidence suggests that aberrant post-translational modifications, including ubiquitination, acetylation and phosphorylation, can lead to aggregation and mislocalization of these proteins, thereby resulting in cellular dysfunction [80]. For instance, TDP-43 is an RNA-binding protein that localizes in the nucleus and regulates a multitude of RNA processing steps, such as transcriptional repression, microRNA biogenesis, pre-mRNA splicing, mRNA stability, mRNA transport and translation. It has been suggested that aggregated TDP-43 shuttles from the nucleus to the cytoplasm, and its accumulation and redistribution to the neurites lead to the loss-of-function of normal TDP-43, thereby affecting cellular metabolism. Alternatively, the presence of cleaved TDP-43 C-terminal fragment(s) (25 and 35 kDa) may result in a toxic gain of function [81]. More recently, it has been shown that neuroinflammation in ALS is driven, in part, by cytoplasmic DNA sensor cyclic guanosine monophosphate (GMP)-AMP synthase (cGAS) resulting from TDP-43-induced mtDNA release. In fact, inhibition of cGAS averts upregulation of NF-κB and type I IFN responses in iPSC-derived motor neurons and in TDP-43 mutant mice [82]. Similarly, FUS is also an RNA-binding protein. It is localized in the nucleus where it regulates RNA processing pathways. In neurons, FUS is present in RNA-transporting granules, axons, dendrites, and excitatory synapses. While the loss of FUS does not trigger motor neuron degeneration and is not sufficient to cause ALS, overexpression of mutant or wild-type FUS triggers motor neuronal cell death, supporting toxic gain of function [76].
In both sporadic and familial ALS, motor neuron cell death occurs by complex mechanisms that involve dysfunction in several cellular processes, including endoplasmic reticulum and mitochondrial stress, autophagy, proteasome, oxidative stress (ROS and nitric oxide), glutamate excitotoxicity, calcium overload, aberrant RNA/DNA regulation, axonal transport system impairments and the “prion-like” propagation of misfolded protein, where the aggregated/abnormal proteins act as seeds of neuropathology [83]. Neuroinflammation is increasingly being recognized as a prominent pathological feature of ALS. Here we briefly review the role of the immune system, especially focusing on the role of the central nervous system (CNS) innate immunity in neurodegeneration, and further discuss potential therapeutic approaches to precisely dampening chronic neuroinflammatory responses in ALS by targeting the NLRP3 inflammasome.
4.1. Neuroinflammation and Motor Neuron Cell Death
Although the etiology and pathogenesis of ALS continues to be debated, accumulating evidence suggests that neuroinflammation is an active contributor to the initiation and progression of ALS [84]. The neuroinflammatory responses are mediated, at least in part, by the activation of resident innate immune cells, i.e., microglia, astrocytes and mast cells, in response to sterile triggers (e.g., protein aggregates). These responses are initially beneficial and protect the host. However, failure to regulate these processes leads to a state of sustained/chronic activation of the immune system, which constitutively secretes pathogenic levels of proinflammatory mediators and can contribute to motor neuron death and disease progression [85][86][87]. Indeed, reactive astrocytes and microglia are generally found to accumulate at sites of motor neuron degeneration and have been shown to release proinflammatory cytokines, including TNF-α, CD11b, IL-17A, interferon gamma (IFNγ), CD40L and IL-6; and NLRP3 inflammasome-induced cytokines IL-1β and IL-18, in post-mortem human tissues and animal models of ALS, respectively [88]. In addition, ALS patients display a detrimental neuroinflammatory phenotype, not only due to increased production of proinflammatory cytokines by activated resident immune cells but also because of increased peripheral immune cell migration into the brain, which can further aggravate inflammatory responses [89]. Increasing evidence points to an extensive and complex interaction occurring between the innate immune cells, i.e., mast cells, astrocytes and microglia, which orchestrate immune responses during neuroinflammation, and neurodegeneration. Several studies have also demonstrated increased infiltration of immune cells from the circulation into the brain; however, it is still unknown whether they are protective or detrimental [90]. Notably, blood–CNS barrier impairments and changes in endothelial cells have been documented in both human pathological samples and animal models of ALS [91].
Current evidence also points to the roles of the peripheral immune system in the initiation and progression of ALS, which have recently been reviewed [92]. A histopathological, post-mortem assessment of human spinal cord and brain tissues revealed T-lymphocyte infiltration at the sites of motor neuron cell death, suggesting engagement of the adaptive immune system [93]. Flow cytometry analysis demonstrated alterations in T-lymphocyte populations in the blood of ALS patients as compared to healthy subjects. Decreases in the regulatory T-lymphocyte (Tregs) cell counts and mRNA levels of FoxP3 (transcription factor forkhead box P3) were associated with accelerated disease progression in ALS patients [94]. In addition, Tregs obtained from ALS patients were less effective at suppressing responder T-lymphocytes as compared to Tregs from healthy subjects, and circulating monocytes of ALS patients expressed a distinct proinflammatory signature [95]. In a human clinical study involving 33 patients with sporadic ALS and 38 healthy control subjects, Sheean et al. recently showed that a systemic reduction in peripheral Tregs (including effector CD45RO+/FoxP3+ population) correlated with an enhanced rate of ALS progression. They also tested the therapeutic potential of enhancing effector Tregs in transgenic mice harboring a G93A mutation in SOD1 (SOD1G43A) [96]. Expansion of effector Tregs population was associated with slower disease progression and improvement in the lifespan for ALS mice. At the histological level, marked reductions in astrogliosis and microgliosis were accompanied by increased motor neuron survival [96]. Tregs are known to suppress inflammation by inhibiting proinflammatory effector T-lymphocytes, and proinflammatory innate immune myeloid cells, such as monocytes and macrophages; and by promoting neuroprotective microglia [94]. Taken together, these studies demonstrate a clear link between patient Tregs populations, monocyte profiles and ALS progression, and support that there is active crosstalk between the innate and adaptive immune systems. Strategies aimed at enhancing Tregs might be immunomodulatory and provide neuroprotection leading to improved clinical outcomes in ALS patients.
A vast number of studies highlight the complexity of the immune responses in ALS patients and provide compelling evidence for immune dysregulation in the pathogenesis of ALS (comprehensively reviewed in [97][98]). There exists a conundrum between hyperinflammation and inefficient immune responses to pathogenic stimuli in ALS patients, possibly due to patient-specific mutations in ALS-associated genes or polymorphisms in cytokine/chemokine genes that might further affect the patient’s ability to mount appropriate immune responses.
4.2. Astrocytes: Stars in the Darkness
Astrocytes represent the most abundant cell type in the CNS that regulate and maintain homeostasis [99]. Such astrocytes are described as “homeostatic astrocytes” which are involved in critical CNS functions, including but not limited to the maintenance and regulation of the extracellular microenvironment, blood–brain barrier (BBB) integrity, cerebral blood flow, antioxidant and trophic factor support, uptake and recycling of neurotransmitters and detoxification of extracellular glutamate and ROS [100][101][102]. Astrocytes respond to all forms of CNS damage, including misfolded aggregated proteins composed of SOD1, FUS and TDP-43; dying neurons; and elevated ROS and cytokines, by a process known as “reactive astrocytosis” which is accompanied by increased expression of intermediate filaments such as vimentin and glial fibrillary acidic protein (GFAP) [103]. Reactive astrocytes also undergo a dramatic morphological transformation and exhibit ramifications of GFAP-positive hypertrophic processes which are associated with a broad spectrum of molecular and functional changes [104]. Liddelow et al. separated reactive astrocytes into A1 and A2 subtypes: A1 astrocytes were shown to be neurotoxic and exacerbate disease pathology, whereas A2 astrocytes secreted neurotrophic factors and promoted neuronal survival [105]. It has now been established that reactive astrocytosis is a highly dynamic process whereby some astrocytes can lose their normal functions and gain neurotoxic functions, which in turn, can negatively impact the surrounding neural tissue whereas others can revert back to their normal homeostatic states and promote neuronal survival. However, identification of the exact molecular subtypes of reactive astrocytes that are beneficial or detrimental to neuronal health has been technically challenging, and their roles in the neurodegenerative process remain elusive.
4.3. Microglia: The Defense System of the CNS
Microglia represent the resident innate immune cells of the CNS that are important for the maintenance of homeostasis and neuronal survival. Like other tissue macrophages, microglia are the professional phagocytes of the CNS that defend against microbial and non-microbial threats and clear debris from damaged/dead cells. As a first line of defense, following detection of neurodegeneration-associated molecular patterns (NAMPs), microglia activate a defense program that arms them with a toolkit to transition from the “homeostatic” state into an “activated” or a “disease-associated” state [106]. During the transition to the activated state, microglia undergo numerous morphofunctional changes, release inflammatory mediators (TNF-α, IL-6, nitric oxide, ROS and superoxide anions) and trigger activation of the NLRP3 inflammasome [107]. Following damage resolution, microglia shift back to the homeostatic state and secrete neuronal survival molecules, such as anti-inflammatory cytokines (IL-10 and IL-4), and neurotrophic growth factors, to ensure neuronal fitness. However, chronic stimulation switches microglia to a hyperinflammatory state, resulting in continuous production of proinflammatory mediators, and leading to motor neuron death. Using high-throughput RNA sequencing, a recent study has shown that microgliosis in ALS is driven by a subpopulation of microglia with transcriptional signatures similar to those of disease-associated microglia [108]. This unique population of microglial cells might play an important role in driving neuroinflammatory responses and synaptic loss seen in ALS disease progression.
In summary, increased understanding of the mechanisms involved in NLRP3 inflammasome formation and activation will lead to the development of effective treatments for neuromuscular diseases.
References
- Miyatake, S.; Shimizu-Motohashi, Y.; Takeda, S.; Aoki, Y. Anti-inflammatory drugs for duchenne muscular dystrophy: Focus on skeletal muscle-releasing factors. Drug Des. Dev. Ther. 2016, 10, 2745–2758.
- Costamagna, D.; Costelli, P.; Sampaolesi, M.; Penna, F. Role of inflammation in muscle homeostasis and myogenesis. Mediat. Inflamm. 2015, 2015, 805172.
- Zhong, Y.; Kinio, A.; Saleh, M. Functions of NOD-like receptors in human diseases. Front. Immunol. 2013, 4, 333.
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832.
- Hamarsheh, S.; Zeiser, R. NLRP3 inflammasome activation in cancer: A double-edged sword. Front. Immunol. 2020, 11, 1444.
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606.
- Zhang, H.; Zahid, A.; Ismail, H.; Tang, Y.; Jin, T.; Tao, J. An overview of disease models for NLRP3 inflammasome over-activation. Expert Opin. Drug Discov. 2020, 16, 1–18.
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S.M. Downregulation of the NLRP3 inflammasome by adiponectin rescues duchenne muscular dystrophy. BMC Biol. 2018, 16, 33.
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489.
- McKee, C.M.; Coll, R.C. NLRP3 inflammasome priming: A riddle wrapped in a mystery inside an enigma. J. Leukoc. Biol. 2020, 108, 937–952.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Duncan, J.A.; Bergstralh, D.T.; Wang, Y.; Willingham, S.B.; Ye, Z.; Zimmermann, A.G.; Ting, J.P.-Y. Cryopyrin/NALP3 binds atp/datp, is an atpase, and requires atp binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 8041–8046.
- He, W.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298.
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75.
- Stehlik, C.; Lee, S.H.; Dorfleutner, A.; Stassinopoulos, A.; Sagara, J.; Reed, J.C. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J. Immunol. 2003, 171, 6154–6163.
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.-W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: A supramolecular assembly of asc dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604.
- Dick, M.S.; Sborgi, L.; Rühl, S.; Hiller, S.; Broz, P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat. Commun. 2016, 7, 11929.
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 1–11.
- Ferrari, D.; Villalba, M.; Chiozzi, P.; Falzoni, S.; Ricciardi-Castagnoli, P.; Di Virgilio, F. Mouse microglial cells express a plasma membrane pore gated by extracellular ATP. J. Immunol. 1996, 156, 1531–1539.
- Ferrari, D.; Pizzirani, C.; Adinolfi, E.; Lemoli, R.M.; Curti, A.; Idzko, M.; Panther, E.; Di Virgilio, F. The P2X7 receptor: A key player in IL-1 processing and release. J. Immunol. 2006, 176, 3877–3883.
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Núñez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343.
- Ratsimandresy, R.A.; Chu, L.H.; Khare, S.; de Almeida, L.; Gangopadhyay, A.; Indramohan, M.; Misharin, A.V.; Greaves, D.R.; Perlman, H.; Dorfleutner, A.; et al. The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat. Commun. 2017, 8, 15556.
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am. J. Pathol. 2010, 176, 2891–2900.
- Bracey, N.A.; Beck, P.L.; Muruve, D.A.; Hirota, S.A.; Guo, J.; Jabagi, H.; Wright, J.R.; Macdonald, J.A.; Lees-Miller, J.P.; Roach, D.; et al. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp. Physiol. 2013, 98, 462–472.
- Liu, Y.; Bi, X.; Zhang, Y.; Wang, Y.; Ding, W. Mitochondrial dysfunction/NLRP3 inflammasome axis contributes to angiotensin II-induced skeletal muscle wasting via PPAR-γ. Lab. Investig. 2020, 100, 712–726.
- Malik, U.; Raizada, V. Some aspects of the renin-angiotensin-system in hemodialysis patients. KBR 2015, 40, 614–622.
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708.
- Li, W.; Moylan, J.S.; Chambers, M.A.; Smith, J.; Reid, M.B. Interleukin-1 stimulates catabolism in C2C12 Myotubes. Am. J. Physiol. Cell Physiol. 2009, 297, C706–C714.
- Llovera, M.; Carbó, N.; López-Soriano, J.; García-Martínez, C.; Busquets, S.; Alvarez, B.; Agell, N.; Costelli, P.; López-Soriano, F.J.; Celada, A.; et al. Different cytokines modulate ubiquitin gene expression in rat skeletal muscle. Cancer Lett. 1998, 133, 83–87.
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in muckle-wells autoinflammatory disorder. Immunity 2004, 20, 319–325.
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748.
- Ankala, A.; Silva, C.d.; Gualandi, F.; Ferlini, A.; Bean, L.J.H.; Collins, C.; Tanner, A.K.; Hegde, M.R. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann. Neurol. 2015, 77, 206–214.
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD global database: Analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402.
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Gappmaier, E.; Howard, M.T.; Sampson, J.B.; Mendell, J.R.; Wall, C.; King, W.M.; et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort. Hum. Mutat. 2009, 30, 1657–1666.
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267.
- Mayer, O.H.; Finkel, R.S.; Rummey, C.; Benton, M.J.; Glanzman, A.M.; Flickinger, J.; Lindström, B.-M.; Meier, T. Characterization of pulmonary function in duchenne muscular dystrophy. Pediatr. Pulmonol. 2015, 50, 487–494.
- Landfeldt, E.; Thompson, R.; Sejersen, T.; McMillan, H.J.; Kirschner, J.; Lochmüller, H. Life expectancy at birth in duchenne muscular dystrophy: A systematic review and meta-analysis. Eur. J. Epidemiol. 2020, 35, 643–653.
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319.
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular cell adhesion molecule-1 expression and signaling during disease: Regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638.
- Hodgetts, S.; Radley, H.; Davies, M.; Grounds, M.D. Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking tnfalpha function with etanercept in mdx mice. Neuromuscul. Disord. 2006, 16, 591–602.
- Savino, W.; Pinto-Mariz, F.; Mouly, V. Flow Cytometry-Defined CD49d Expression in Circulating T-Lymphocytes Is a Biomarker for Disease Progression in Duchenne Muscular Dystrophy. Methods Mol. Biol. 2018, 1687, 219–227.
- Guiraud, S.; Davies, K.E. Pharmacological advances for treatment in duchenne muscular dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48.
- Vitiello, L.; Tibaudo, L.; Pegoraro, E.; Bello, L.; Canton, M. Teaching an old molecule new tricks: Drug repositioning for duchenne muscular dystrophy. Int. J. Mol. Sci. 2019, 20, 6053.
- Barnes, P.J. How corticosteroids control inflammation: Quintiles prize lecture 2005. Br. J. Pharmacol. 2006, 148, 245–254.
- Serra, F.; Quarta, M.; Canato, M.; Toniolo, L.; De Arcangelis, V.; Trotta, A.; Spath, L.; Monaco, L.; Reggiani, C.; Naro, F. Inflammation in muscular dystrophy and the beneficial effects of non-steroidal anti-inflammatory drugs. Muscle Nerve 2012, 46, 773–784.
- Péladeau, C.; Adam, N.J.; Jasmin, B.J. Celecoxib treatment improves muscle function in mdx mice and increases utrophin a expression. FASEB J. 2018, 32, 5090–5103.
- Henriques-Pons, A.; Yu, Q.; Rayavarapu, S.; Cohen, T.V.; Ampong, B.; Cha, H.J.; Jahnke, V.; Van der Meulen, J.; Wang, D.; Jiang, W.; et al. Role of toll-like receptors in the pathogenesis of dystrophin-deficient skeletal and heart muscle. Hum. Mol. Genet. 2014, 23, 2604–2617.
- Torres, L.F.; Duchen, L.W. The mutant mdx: Inherited myopathy in the mouse. morphological studies of nerves, muscles and end-plates. Brain 1987, 110, 269–299.
- Briguet, A.; Courdier-Fruh, I.; Foster, M.; Meier, T.; Magyar, J.P. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul. Disord. 2004, 14, 675–682.
- Abou-Samra, M.; Lecompte, S.; Schakman, O.; Noel, L.; Many, M.C.; Gailly, P.; Brichard, S.M. Involvement of adiponectin in the pathogenesis of dystrophinopathy. Skelet. Muscle 2015, 5, 1–17.
- Baatar, D.; Patel, K.; Taub, D.D. The Effects of ghrelin on inflammation and the immune system. Mol. Cell Endocrinol. 2011, 340, 44–58.
- Huang, C.-X.; Yuan, M.-J.; Huang, H.; Wu, G.; Liu, Y.; Yu, S.-B.; Li, H.-T.; Wang, T. Ghrelin inhibits post-infarct myocardial remodeling and improves cardiac function through anti-inflammation effect. Peptides 2009, 30, 2286–2291.
- Sun, G.-X.; Ding, R.; Li, M.; Guo, Y.; Fan, L.-P.; Yue, L.-S.; Li, L.-Y.; Zhao, M. Ghrelin attenuates renal fibrosis and inflammation of obstructive nephropathy. J. Urol. 2015, 193, 2107–2115.
- Chang, L.; Niu, F.; Chen, J.; Cao, X.; Liu, Z.; Bao, X.; Xu, Y. Ghrelin improves muscle function in dystrophin-deficient mdx mice by inhibiting NLRP3 inflammasome activation. Life Sci. 2019, 232, 116654.
- Moresi, V.; Adamo, S.; Berghella, L. The JAK/STAT pathway in skeletal muscle pathophysiology. Front. Physiol. 2019, 10, 500.
- Lau, Y.S.; Zhao, L.; Zhang, C.; Li, H.; Han, R. Genetic disruption of the inflammasome adaptor ASC has minimal impact on the pathogenesis of duchenne muscular dystrophy in mdx mice. Life Sci. 2020, 257, 118069.
- Bushby, K.M.; Beckmann, J.S. The limb-girdle muscular dystrophies-proposal for a new nomenclature. Neuromuscul. Disord. 1995, 5, 337–343.
- Wicklund, M.P.; Kissel, J.T. The limb-girdle muscular dystrophies. Neurol. Clin. 2014, 32, 729–749.
- Di Fruscio, G.; Garofalo, A.; Mutarelli, M.; Savarese, M.; Nigro, V. Are all the previously reported genetic variants in limb girdle muscular dystrophy genes pathogenic? Eur. J. Hum. Genet. 2016, 24, 73–77.
- Liewluck, T.; Milone, M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve 2018, 58, 167–177.
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.-C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172.
- Han, R. Muscle membrane repair and inflammatory attack in dysferlinopathy. Skelet. Muscle 2011, 1, 10.
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473.
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.W.; Van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria mediate cell membrane repair and contribute to duchenne muscular dystrophy. Cell Death Differ. 2017, 24, 330–342.
- Shkryl, V.M.; Martins, A.S.; Ullrich, N.D.; Nowycky, M.C.; Niggli, E.; Shirokova, N. Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflüg. Arch. 2009, 458, 915–928.
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 2005, 562, 367–380.
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287.
- McDade, J.R.; Michele, D.E. Membrane damage-induced vesicle–vesicle fusion of dysferlin-containing vesicles in muscle cells requires microtubules and kinesin. Hum. Mol. Genet. 2014, 23, 1677–1686.
- Kinnunen, K.; Piippo, N.; Loukovaara, S.; Hytti, M.; Kaarniranta, K.; Kauppinen, A. Lysosomal destabilization activates the NLRP3 inflammasome in human umbilical vein endothelial cells (HUVECs). J. Cell Commun. Signal. 2017, 11, 275–279.
- Manna, S.; Howitz, W.J.; Oldenhuis, N.J.; Eldredge, A.C.; Shen, J.; Nihesh, F.N.; Lodoen, M.B.; Guan, Z.; Esser-Kahn, A.P. Immunomodulation of the NLRP3 inflammasome through structure-based activator design and functional regulation via lysosomal rupture. ACS Cent. Sci. 2018, 4, 982–995.
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955.
- Hulisz, D. Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 2018, 24, S320–S326.
- Gros-Louis, F.; Gaspar, C.; Rouleau, G.A. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 956–972.
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794.
- Umoh, M.E.; Fournier, C.; Li, Y.; Polak, M.; Shaw, L.; Landers, J.E.; Hu, W.; Gearing, M.; Glass, J.D. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology 2016, 87, 1024–1030.
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13.
- Rosen, D.R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362.
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; et al. Mutation of SOD1 in ALS: A gain of a loss of function. Hum. Mol. Genet. 2007, 16, 1604–1618.
- Yamashita, S.; Ando, Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 13.
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45.
- Forman, M.S.; Trojanowski, J.Q.; Lee, V.M.-Y. TDP-43: A novel neurodegenerative proteinopathy. Curr. Opin. Neurobiol. 2007, 17, 548–555.
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 triggers mitochondrial DNA release via MPTP to activate CGAS/STING in ALS. Cell 2020, 183, 636–649.e18.
- Hayashi, Y.; Homma, K.; Ichijo, H. SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv. Biol. Regul. 2016, 60, 95–104.
- Liu, J.; Wang, F. Role of neuroinflammation in amyotrophic lateral sclerosis: Cellular mechanisms and therapeutic implications. Front. Immunol. 2017, 8, 1005.
- Deora, V.; Lee, J.D.; Albornoz, E.A.; McAlary, L.; Jagaraj, C.J.; Robertson, A.A.B.; Atkin, J.D.; Cooper, M.A.; Schroder, K.; Yerbury, J.J.; et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 2020, 68, 407–421.
- Morgan, S.; Orrell, R.W. Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 2016, 119, 87–98.
- Trias, E.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Kovacs, M.; Moura, I.C.; Beckman, J.S.; Hermine, O.; et al. Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight 2018, 3.
- Michaelson, N.; Facciponte, D.; Bradley, W.; Stommel, E. Cytokine expression levels in ALS: A potential link between inflammation and BMAA-triggered protein misfolding. Cytokine Growth Factor Rev. 2017, 37, 81–88.
- Komine, O.; Yamanaka, K. Neuroinflammation in motor neuron disease. Nagoya J. Med. Sci. 2015, 77, 537–549.
- Herndon, J.M.; Tome, M.E.; Davis, T.P. Chapter 9—Development and maintenance of the blood-brain barrier. In Primer on Cerebrovascular Diseases, 2nd ed.; Caplan, L.R., Biller, J., Leary, M.C., Lo, E.H., Thomas, A.J., Yenari, M., Zhang, J.H., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 51–56. ISBN 9780128030585.
- Garbuzova-Davis, S.; Sanberg, P.R. Blood-CNS barrier impairment in ALS patients versus an animal model. Front. Cell Neurosci. 2014, 8, 21.
- McCombe, P.A.; Lee, J.D.; Woodruff, T.M.; Henderson, R.D. The peripheral immune system and amyotrophic lateral sclerosis. Front. Neurol. 2020, 11, 279.
- Graves, M.C.; Fiala, M.; Dinglasan, L.A.V.; Liu, N.Q.; Sayre, J.; Chiappelli, F.; van Kooten, C.; Vinters, H.V. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and t cells. Amyotroph. Lateral Scler. Other Mot. Neuron Dis. 2004, 5, 213–219.
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory t-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2013, 5, 64–79.
- Beers, D.R.; Zhao, W.; Wang, J.; Zhang, X.; Wen, S.; Neal, D.; Thonhoff, J.R.; Alsuliman, A.S.; Shpall, E.J.; Rezvani, K.; et al. ALS patients’ regulatory t-lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight 2017, 2, e89530.
- Sheean, R.K.; McKay, F.C.; Cretney, E.; Bye, C.R.; Perera, N.D.; Tomas, D.; Weston, R.A.; Scheller, K.J.; Djouma, E.; Menon, P.; et al. Association of regulatory t-cell expansion with progression of amyotrophic lateral sclerosis: A study of humans and a transgenic mouse model. JAMA Neurol. 2018, 75, 681–689.
- Béland, L.-C.; Markovinovic, A.; Jakovac, H.; De Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in amyotrophic lateral sclerosis: Blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2020, 2, fcaa124.
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220.
- Miller, S.J. Astrocyte heterogeneity in the adult central nervous system. Front. Cell. Neurosci. 2018, 12.
- Banker, G.A. Trophic interactions between astroglial cells and hippocampal neurons in culture. Science 1980, 209, 809–810.
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686.
- Koehler, R.C.; Roman, R.J.; Harder, D.R. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009, 32, 160–169.
- Kushner, P.D.; Stephenson, D.T.; Wright, S. Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. J. Neuropathol. Exp. Neurol. 1991, 50, 263–277.
- Pekny, M.; Nilsson, M. Astrocyte activation and reactive gliosis. Glia 2005, 50, 427–434.
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487.
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-associated microglia: A universal immune sensor of neurodegeneration. Cell 2018, 173, 1073–1081.
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477.
- Dols-Icardo, O.; Montal, V.; Sirisi, S.; López-Pernas, G.; Cervera-Carles, L.; Querol-Vilaseca, M.; Muñoz, L.; Belbin, O.; Alcolea, D.; Molina-Porcel, L.; et al. Motor cortex transcriptome reveals microglial key events in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7.


