Adult-onset Still’s disease (AOSD) is a non-familial, polygenic systemic autoinflammatory disorder. It is traditionally characterized by four cardinal manifestations—spiking fever, an evanescent salmon-pink maculopapular rash, arthralgia or arthritis and a white-blood-cell count (WBC) ≥ 10,000/mm3, mainly neutrophilic polymorphonuclear cells (PMNs)—but many other manifestations and complications can be associated, making clinical expression very heterogeneous and diagnosis sometimes difficult. The AOSD course can be diverse and is currently impossible to predict. Several clinical phenotypes have been described, either on the basis of the evolution of symptoms over time (monocyclic, polycyclic and chronic evolution) or according to dominant clinical evolution (systemic and arthritis subtypes). However, these patterns are mainly based on case series and not on robust epidemiological studies. Furthermore, they have mainly been established a long time ago, before the era of the biological treatments.
1. Introduction
Adult-onset Still’s disease (AOSD) is classically described as a non-familial, or sporadic, systemic autoinflammatory disorder (SAID)
[1]. Its incidence is estimated at 0.16 to 0.4 per 100,000 persons
[2][3], and the reported prevalence rates range from 1 to 34 cases per 1 million persons in the Japanese and the European populations
[3]. It is traditionally characterized by four cardinal manifestations—spiking fever, an evanescent salmon-pink maculopapular rash, arthralgia or arthritis and a white-blood-cell count (WBC) ≥ 10,000/mm
3, mainly neutrophilic polymorphonuclear cells (PMNs)—but many other manifestations and complications can be associated, making clinical expression very heterogeneous
[2] and diagnosis sometimes difficult. Classification criteria can be of help to perform the latter
[4][5]. The AOSD course can also be diverse and is currently impossible to predict
[6]. Several phenotypes have been described; however, these phenotypes are mainly based on case series and not on robust epidemiological studies
[7][8][9][10][11]. Furthermore, they have mainly been established a long time ago, before the era of the biological treatments. In the first part of this paper, we will describe the “typical” presentation of AOSD and the evolution courses described so far. In the second part, based on our personal experience and on recent advances in the understanding of disease pathogenesis, we will discuss why it appears interesting to reshuffle AOSD phenotypes, emphasizing the continuum between AOSD profiles and with other systemic autoinflammatory disorders.
2. Common and Rare Clinical Manifestations
Besides the cardinal manifestations listed above, many other frequent clinical manifestations or biological abnormalities can exist in AOSD
[1][2] and are recalled in the second section of Table 1. AOSD can also be responsible for a number of serious and life-threatening complications
[12] (last section of Table 1). The physician should be aware of these atypical forms because the intensity of the general signs or organ involvement often relegates the cardinal signs to the background. The diagnosis of AOSD is then difficult and often delayed, after many other diagnostic hypotheses have been excluded.
Table 1. Possible manifestations of AOSD.
| Cardinal Manifestations |
| Skin rash |
| Fever > 39 °C |
| Leukocytes > 10,000/mm3, neutrophils > 80% |
| Arthritis and arthralgia |
| Other frequent manifestations |
| Odynophagia, pharyngitis |
| Myalgia, myositis |
| Lymphadenopathy, splenomegaly |
| Hepatomegaly, hepatitis |
| Pericarditis, myocarditis, pleuritis, lung disease (interstitial lung infiltrates) |
| Increased ESR, CRP, fibrinogen |
| Increased ferritin, decreased glycosylated ferritin |
| Life-threatening complications |
| Tamponade, myocarditis, and acute respiratory syndrome |
| Pulmonary arterial hypertension |
| Fulminant hepatitis |
| Macrophage activation syndrome |
| Disseminated intravascular coagulopathy |
| Thrombotic microangiopathy |
After comprehensive investigations, no clinical sign or biological abnormality is specific to AOSD to ascertain diagnosis. The classification criteria may be helpful, but these criteria were developed for clinical research rather than for diagnosis. Two sets of criteria have been validated. The Yamaguchi criteria, published in 1992, are the most widely used
[4] (Table 2). These criteria include exclusion criteria such as infections, malignancies and other rheumatic diseases. They should be used only after a broad diagnostic work-up, which is problematic in clinical practice. The Fautrel criteria have the advantage of including ferritin and glycosylated ferritin (GF) levels as diagnostic biomarkers and do not require exclusion criteria
[5] (Table 2). In a 2018 validation study, both sets showed high sensitivity and specificity
[13].
Table 2. Classification criteria for AOSD.
| Criteria |
Yamaguchi et al. [4] |
Fautrel et al. [5] |
| Major criteria |
Fever ≥ 39 °C lasting one week or more
Arthralgia lasting two weeks or more
Typical skin rash: maculopapular, non-pruritic, salmon-pink rash with concomitant fever spikes
Leukocytosis ≥ 10,000/mm3 with neutrophil polymorphonuclear proportion ≥ 80% |
Spiking fever ≥ 39 °C
Arthralgia
Transient erythema
Pharyngitis
Neutrophil polymorphonuclear
proportion ≥ 80%
GF proportion ≤ 20% |
| Minor criteria |
Pharyngitis or sore throat
Lymphadenopathy and/or splenomegaly
Liver enzyme abnormalities (aminotransferases)
Negative for RF or antinuclear antibodies |
Typical rash
Leukocytosis ≥ 10,000/mm3 |
| Exclusion criteria |
Absence of infection, especially sepsis and
Epstein–Barr viral infection
Absence of malignant diseases, especially
Lymphomas
Absence of inflammatory disease, especially polyarteritis nodosa |
None |
| Criteria requirement |
At least five criteria, including two major criteria and no exclusion criteria |
Four major criteria or three major criteria and two minor criteria |
| Set performance |
Sensitivity 96.3%, specificity 98.2%, PPV 94.6% and NPV 99.3%
Modified Yamaguchi criteria, i.e., Yamaguchi criteria and ferritin > ULN: sensitivity 100%, specificity 97.1%, PPV 87.1% and NPV 100%
Alternative modified Yamaguchi criteria, i.e., Yamaguchi criteria and GF ≤ 20%: sensitivity 98.2%, specificity 98.6%, PPV 93.0% and NPV 99.6% [13] |
Sensitivity 87.0%, Specificity 97.8%, PPV 88.7% and NPV 97.5% [13] |
3. Evolution Courses
Classically, AOSD phenotypes and evolution courses have been described based on two different aspects: the evolution of symptoms over time (monocyclic, polycyclic and chronic) and/or the type of symptoms, i.e., systemic (recurrent fever syndrome) versus articular (febrile, potentially erosive polyarthritis).
3.1. Classification Based on the Evolution Temporality
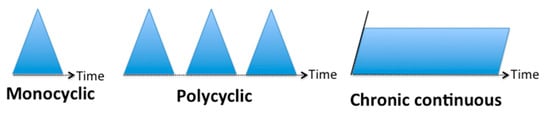
Historically, the clinical course of AOSD has been distinguished into three different phenotypes, described on the basis of the evolution of symptoms over time: monocyclic, polycyclic and chronic evolution (Figure 1)
[1].
Figure 1. AOSD phenotypes based on the evolution of symptoms over time.
A monocyclic course is either self-limited or includes drug-free remission. The initial flare up with systemic manifestations and (potentially) joint involvement develops over a few weeks. The disease is limited to a single flare up and complete remission is achieved within a couple of weeks or months. Remission can be achieved with nonsteroidal inflammatory drugs, steroids or other immunomodulatory agents after a few days or weeks. These treatments can be progressively tapered then stopped without relapse after a few months. This phenotype seems to account for 19 to 44% of affected patients
[9][10][14].
A recurrent or polycyclic course is characterized by AOSD relapse following either months or years of immunomodulatory treatment or therapy discontinuation. In this pattern, one possible presentation is a first flare up during systemic juvenile idiopathic arthritis (SJIA) diagnosed in childhood, followed by sustained drug-free remission for several years then a relapse in adulthood. In most of these cases, recurrences combine systemic manifestations and joint involvement. This phenotype represents 10 to 41% of affected patients
[9][10][14].
Differently from the others, a chronic and progressive course involves continuous inflammation that is responsible for chronic erosive joint involvement with regular systemic flares. This phenotype is the most frequent, estimated at 35 to 67% of affected patients, in old series from the pre-biologic era
[9][11][14][15].
3.2. Classification Based on the Type of Symptoms
In light of the new pieces of evidence about AOSD pathogenesis and treatment, especially the current use of biological therapy, this historic classification seems somehow outdated.
Many authors have for some time now adopted a dichotomous classification, distinguishing two AOSD subtypes: systemic and articular. The systemic subtype includes patients with systemic features (such as high fever and skin rash), more at risk to develop life-threatening complications (such as multi-organ involvement and RHL). In the articular subtype patients have predominant articular involvement
[16][17][18][19]. Classical articular manifestations described in AOSD are varied. Joint pain is the most common symptom, usually starting concomitantly with fever and with a maximal intensity during fever spikes. Arthritis is present in more than two-thirds of patients, migrating at the very beginning and becoming stable in the course of the disease. Bilateral symmetrical rheumatoid arthritis (RA)-like polyarthritis has been described. Any joint can be involved, more frequently radio-carpal or carpal joints (with the “classical bilateral ankylosing carpal arthritis”, which is very suggestive when “isolated”, i.e., without concomitant metacarpo-phalangeal or proximal interphalangeal structure damage)
[1][2][9][11][15].
Predictive factors for the evolution towards each subset have been identified: high fever (>39 °C), hepatitis, thrombocytopenia, elevated CRP and hyperferritinemia seem to be associated with a systemic subset, while female gender, proximal arthritis at disease onset and steroid dependence are predictive of a chronic articular evolution
[16][17][18][19][20][21].
3.3. Pathogenic Consideration and Expected Therapeutic Outcomes
Substantial advances have been made to confirm the homology between AOSD and systemic-onset juvenile arthritis (SJIA, formerly called childhood-onset Still’s disease), and currently, most authors believe that they correspond to the same sporadic systemic auto-inflammatory disorder associated with inappropriate activation of the innate immune system at different ages
[1]. It is thought to be a complex autoinflammatory interleukin (IL)-1-mediated disease, with IL-6- and IL-18-mediated inflammatory features as well.
Data from the literature suggest that different cytokine profiles may be responsible for distinct clinical manifestations, as systemic subsets seem to present with high levels of IL-18 and IL-1β while patients with arthritis exhibit higher IL-6 serum levels
[16][20]. In a cohort comparing 33 AOSD cases with 77 SJIA cases, patients with AOSD were classified into two subgroups based on serum IL-6 and IL-18 levels. The number of patients with arthritis was significantly higher in the IL-6-dominant subgroup, while no patient in the IL-18-dominant subgroup presented with arthritis
[20]. However, these data should be treated with caution since “cytokine profiles” have not been clearly established. While IL-6 serum levels are higher in patients with arthritis, this cytokine has also been associated with severe systemic manifestations such as macrophage activation syndrome
[17], and therapeutic blockade of IL-6 provided excellent clinical responses in SJIA patients with systemic phenotype
[22].
Consequently, serum levels of cytokines are not routinely performed and do not highlight our daily management of individual patients. They remain to be validated in further prospective studies comparing “systemic” AOSD with “rheumatic” AOSD patients, in order to help “monitoring” the disease and eventually have an impact on therapeutic responses
[6].
 +1 point
+1 point