Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Leo Quinlan | + 2320 word(s) | 2320 | 2021-05-08 12:37:30 | | | |
| 2 | Catherine Yang | Meta information modification | 2320 | 2021-07-07 03:16:47 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Quinlan, L. Modelling Parkinson’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/11738 (accessed on 07 February 2026).
Quinlan L. Modelling Parkinson’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/11738. Accessed February 07, 2026.
Quinlan, Leo. "Modelling Parkinson’s Disease" Encyclopedia, https://encyclopedia.pub/entry/11738 (accessed February 07, 2026).
Quinlan, L. (2021, July 06). Modelling Parkinson’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/11738
Quinlan, Leo. "Modelling Parkinson’s Disease." Encyclopedia. Web. 06 July, 2021.
Copy Citation
Parkinson’s Disease (PD) is a chronic neurodegenerative disorder characterized by motor and non-motor symptoms, among which are bradykinesia, rigidity, tremor as well as mental symptoms such as dementia.
Parkinson’s disease
induced pluripotent stem cells
human pathology
1. Introduction
Parkinson’s disease (PD) is a complex, progressive neurological disorder characterized by degeneration of dopaminergic neurons (DA) in the substantia nigra pars compacta of the ventral mesencephalon [1]. The prevalence of PD is increasing with more than 6.1 million individuals reported globally to have PD in 2016 compared with 2.5 million in 1990 [2]. The increase in prevalence is due both to the ageing global population and associated changes in population behaviors such as smoking, decreased physical activity, and environmental factors such as air pollution [3][4]. PD occurrence is also sex-dependent with males displaying 1.5 times higher incidence compared to females [5]. The characteristic symptoms of PD are generally classified as motor (tremor, bradykinesia, and postural instability) and non-motor (dementia, depression, anxiety, fatigue, and pain) [6][7]. While the majority of PD cases are idiopathic without any clear family history, numerous genetic mutations have been found in individuals, with more rare and familial forms of PD also reported [8]. These genetic factors include autosomal dominant and recessive genes such as leucine-rich-repeat kinase 2 (LRRK2), PARK2 (encoding Parkin), PTEN-induced putative kinase (PINK1), PARK7 (encoding DJ-1), SNCA (encoding α-synuclein), and glucosidase beta acid (GBA). Each of these gene can be seen to present with variable clinical phenotypes.
2. Modelling PD Using iPSCs
There has been remarkable advances in the discovery of genetic mutations associated with PD in recent years [9]. Familial PD collectively accounts for only 10% of all PD cases while the remainder of PD cases have unknown etiology [10][11]. Patient-specific iPSC-derived DA neurons with specific mutations allows the underlying mechanisms of a particular mutation to be investigated in depth. Here, we report on the available data based only on human iPSC-derived neuronal models, investigating specific mutation-associated phenotypes of PD.
2.1. iPSC Modelling of SNCA Mutation and Associated Phenotypes
The first reported mutation associated with autosomal-dominant PD was that of α-synuclein [12]. α-synuclein is involved in numerous important cellular functions such as modulation of intracellular vesicle trafficking [13], dopamine metabolism [14], microtubule nucleation and proliferation [15], as well as the regulation of synaptic vesicle recycling [16]. There are numerous specific mutations associated with SNCA-related PD pathology including A30P, G51D, E46K, A53T, and A53E [17][18][19][20]. The severity of symptoms for PD is proportional to the number of SNCA copies deleted. In human iPSC-derived neurons the most commonly studied alterations in SNCA are the A53T mutation and multiplication of SNCA as they are the most common mutations associated with PD. Alterations in α-synuclein physiology results in a myriad of cellular changes must commonly mediated through mitochondrial dysfunction and oxidative stress. These changes and abnormal aggregation of α-synuclein not only induce neuronal loss but are also seen to prevent neuronal regeneration.
2.2. iPSC Modelling of LRKK2 and Associated Phenotypes
LRRK2 is a multi-domain protein exhibiting both kinase and GTPase functions located in the PARK8 locus on chromosome 12 [21]. LRRK2 is implicated as a significant genetic contributor to the development of autosomal dominant familial PD as well as some cases of idiopathic PD [22][23]. To date around 20 different LRRK2 mutations have been linked to PD pathophysiology [24]. LRRK2 is susceptible to several missense mutations including the LRRK2 G2019S, I2020T, Y1699C, and R1441C heterozygous mutations [25][26]. G2019S is the most commonly occurring mutation and is associated with 4% of familial PD and 1% of idiopathic PD cases [27]. LRKK2 is recognized to have pleiotropic roles across multiple domains including neurite outgrowth [28], modulation of synaptic vesicle endocytosis [29][30] and mitochondrial function and mitophagy [31][32].
2.3. iPSC Modelling of PARK2 Mutations and Associated Phenotypes
Parkin is an E3 ubiquitin ligase, residing in the cytosol, that functions in the ubiquitin proteasome pathway [33][34]. PARK2 which is located on the 6q25.2–27 chromosome encodes parkin and is the most frequent gene mutation associated with autosomal recessive early onset familial PD [34]. Fifty percent of all PD cases under the age of 45 are associated with parkin mutations [35] (Table 1). Mutations in parkin range from small deletions and base pair substitution to large deletions spanning hundreds of nucleotides [36]. Parkin has been primarily shown to be important in maintaining normal mitochondrial function and integrity [37].
PARKIN Mutations Result in Mitochondrial Dysfunction and Oxidative Stress in iPSC-Derived Neurons
Mitochondrial dysfunction, abnormal morphology, and impaired mitochondrial homeostasis are some of the key features displayed by parkin iPSC-derived DA neurons. These neurons exhibit swollen cristae and a highly condensed matrix in the inner mitochondrial membrane (IMM), with abnormal mitochondrial morphology directly affecting function [38] as well as an elevation in the number of enlarged mitochondria [39][40]. Pyruvate kinase M and 14-3-3 epsilon are among the most dysregulated mitochondrial proteins associated with parkin mutated neurons, and this pair have also been consistently shown to be changed in post mortem brain tissues of PD patients [40][41][42].
The culmination of mitochondrial dysfunction is thought to be an increase in oxidative stress, leading to dopamine oxidation [43]. Under normal physiological conditions the transcription of mitochondrial enzymes such as monoamine oxidases (MAO) A and B are limited, as parkin suppresses dopamine-induced oxidative stress [44][45]. However, the levels of MAO-A and B were found to be significantly increased in parkin mutated iPSC-derived DA neurons, suggesting an escalation in dopamine-induced oxidative stress [45]. Increased oxidative stress is a consistent factor observed in numerous independent studies of parkin mutations [38][39][46]. Furthermore, anti-oxidative proteins are significantly reduced in parkin mutated iPSC-derived DA neurons [47]. Conversely, Nrf2, a protein promoting antioxidant gene expression is significantly enhanced in parkin mutated iPSC-derived neurons [38]. Related to this oxidative stress inducing environment, there is a marked decrease in dopamine uptake and increased levels of spontaneous dopamine release in iPSC-derived DA neurons with parkin mutations [48]. This increased spontaneous leak of dopamine is observed in both heterozygous and homozygous forms of parkin neurons, independent of intracellular Ca2+ [48] (Figure 1). Additionally, the number of correctly folded and trafficked dopamine transporters (DAT) are significantly decreased [48]. Thus, parkin is presumed to mitigate dopamine oxidation and control the transmission of dopamine. A further study has demonstrated that the activation of dopamine D1-class receptors in parkin neurons leads to large rhythmic outbursts of spontaneous excitatory postsynaptic currents (sEPSCs) [49] (Figure 1). These rhythmic outburst of sEPSCs resemble oscillatory activities observed within basal ganglia neurons in PD. Overexpression of parkin in this same study resulted in a significant rescue of iPSC-derived neurons, returning oscillatory activities to normal levels [49]. These data show that parkin mutations enhance abnormal dopaminergic modulation and release in neurons.
Table 1. PARK2-mutated iPSC-derived neuronal phenotypes.
| Reference | Number of Cohorts | Type of Mutation | Cell Type | Phenotype |
|---|---|---|---|---|
| [46] | 6 PD patient lines vs. 3 control lines | Exon 2–4 or 6–7 deletions | iPSC-derived DA neurons | 1. Increased Oxidative stress 2. Mitophagy impairment |
| [49] | 3 PD patient lines vs. 3 control lines | Exon 3–5 or R42P deletions | iPSC-derived DA neurons | 1. Dopamine dysregulation |
| [38] | 2 PD patient lines vs. 2 control lines | Exon 2–4 or Exon 6–7 deletions | iPSC-derived DA neurons | 1. Increased oxidative stress 2. Mitochondrial dysfunction 3. Increase α-synuclein aggregation |
| [50] | 4 PD patient lines with 1 control line | Exon 3–4, R275W or R42P deletions | iPSC-derived DA neurons | 1. Mitochondrial dysfunction 2. Increase α-synuclein aggregation |
| [51] | 1 PD patient line vs. 1 control line | Del202-203AG and IVS1+1G/A | iPSC-derived DA neurons | 1. Increased cell death |
| [40] | 2 Isogenic mutated PD lines vs. 1 control line | Exon 2 deletion | iPSC-derived DA neurons | 1. Mitochondrial dysfunction |
| [45] | 2 PD lines vs. 2 control lines | Exon 4 deletion | iPSC-derived DA neurons | 1. Dopamine dysregulation 2. Increased oxidative stress |
| [52] | 2 PD lines vs. 2 control lines | Exon 2–4 deletion | iPSC-derived DA neurons | 1. Mitochondrial dysfunction |
| [53] | 2 Isogenic mutated iPSC lines vs. 1 control line | Exon 2 deletion | iPSC-derived DA neurons | 1. Lysosomal dysfunction |
| [47] | 1 PD line vs. 1 control line | Exon 5 deletion | iPSC-derived DA neurons | 1. Increase α-synuclein aggregation 2. Reduced level of anti-oxidative proteins |
| [54] | 3 PD lines vs. 3 control lines | Exon 7 deletion, c.1072delT or Exon 1 deletion and c.924C>Tor c.1072delT | iPSC-derived DA neurons | 1. Mitochondrial dysfunction |
| [39] | 2 PD lines vs. 2 control lines | c.1366C.T and c.1072Tdel | iPSC-derived DA neurons | 1. Dopamine dysregulation 2. Increase cell death 3. Increase α-synuclein aggregation |
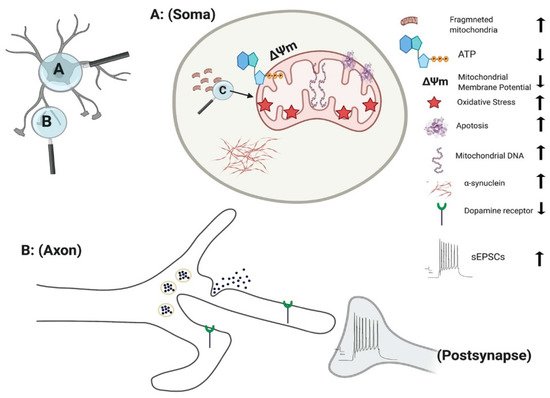
Figure 1. Effect of PINK1 and parkin mutation in IPSC-derived neurons. Parkin mutation in iPSC-derived neurons showed impairment in their requitement to PINK1, alteration in spontaneous postsynaptic current activity, reduction in dopamine receptor and release. PINK1 mutation in iPSC-derived neurons displayed fragmented mitochondria with alteration their DNA level, ATP, membrane potential and oxidative stress. Additionally, there is an increase in apoptotic cell death and α-synuclein aggregation and accumulation.
2.4. iPSC Modelling of PINK1 Mutations and Associated Phenotypes
Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) mutations are the second most frequent cause of early-onset PD and is involved in an autosomal recessive familial form of PD [55]. PINK1-related PD usually appears in the third or fourth decade of life, and like other recessive early onset forms, presents as a slow progression of the disease with a consistent response to levodopa treatment [56]. Forty-two different mutations have been found within the exons of the PINK1 in both heterozygous and homozygous states with Q456X is the most prevalent form of PINK1-related PD mutation [57][58] (Table 2). PINK1 consists of a C-terminal kinase domain and a mitochondrial targeting sequence at the N-terminus. Cytosolic PINK1 is released by truncation of the N-terminal portion of the gene in a proteasome-dependent manner [59][60]. PINK1 has been shown to have an essential role in mitochondrial function, calcium homeostasis, autophagy/mitophagy, protection from stress, and protein misfold [61][62][63].
PINK1 Mutations Result in Loss of Mitochondrial Function and Increases Reactive Oxygen Species Generation in iPSC-Derived Neurons
PINK1 mutations are thought to impair mitochondrial function due to a loss of function, based on the upregulation of PGC-1α and an increase in mitochondrial DNA (mtDNA) copy number (Figure 1) [64]. Under normal physiological conditions, mitochondrial damage activates PINK1 kinase activity, and activated PINK1 phosphorylates ubiquitin at a conserved residue of Ser65 [65]. Parkin cooperates with PINK1 in the phosphorylation process, preparing the damaged mitochondria for lysosomal and proteasomal targeted degradation [66]. Pathogenic mtDNA mutations are found widely in individuals with PD, resulting in mitochondrial dysfunction. Siebler et al., demonstrated that while the level of mtDNA is decreased in wild-type neurons, it remained unchanged in PINK1−/− iPSC-derived neurons upon mitochondrial depolarization, suggesting an increase accumulation of mitochondria DNA dues to loss of PINK function [64]. PINK1+/− iPSC-derived neurons show a significant number of cells with fragmented mitochondria suggestion an alteration in mitochondrial cycling dynamics towards increased organelle fission [67] (Figure 1).
In addition, iPSC-derived neurons with PINK1 mutations show a significant reduction in the level of endogenous parkin levels and are unable to initiate mitophagy due to dysfunction in ubiquitination pathways [68]. Furthermore, the level of Phosphorylated-Ser65-Ub signals are significantly reduced within iPSC-derived TH+ neurons with PINK1 p.G411S mutation [52][69]. Consistent with this, the recruitment of parkin to mitochondria is impaired upon depolarization of mitochondria in PINK1 mutated iPSC-derived DA neurons [64]. Overexpression of wild type PINK1 in these DA neurons restored the translocation of parkin to mitochondria [64]. These studies highlighted the vital role of PINK1 in mitochondrial function and pathogenesis of PD.
One of the major indicators of mitochondrial dysfunction is the generation of reactive oxygen species (ROS) which in turn leads to cell damage due to oxidative stress [70]. Significant damage to lipids, proteins, and nucleic acids have been identified in fibroblast of s carrying PINK mutation [71]. Further study demonstrated that PINK1 deficiency results in an increased basal ROS in both the mitochondria (Figure 1) and cytoplasm, leading to increased oxidative stress in iPSC-derived DA neurons [72]. One of the key mechanisms in detoxification and prevention of ROS associated mitochondrial damage in the cytoplasm is the oxidation of glutathione (GSH). iPSC-derived neurons with PINK1 Q456X mutation display reduced GSH levels and showed increase vulnerability after exposure to low concentrations of valinomycin, concanamycin A, MPP+ and hydrogen peroxide (all promotors of oxidative stress) in comparison to control neurons [73].
Table 2. PINK1-mutated iPSC-derived neuronal phenotypes.
| Reference | Number of Cohorts | Type of Mutation | Cell Type | Phenotype |
|---|---|---|---|---|
| [64] | 3 PD lines vs. 1 control line | c.1366C>T, c.509T>G | iPSC-derived DA neurons | 1. Mitochondrial dysfunction |
| [73] | 5 PD lines vs. 2 control lines | Q456X, R1441C | iPSC-derived DA neurons | 1. Increase in oxidative stress 2. Mitochondrial dysfunction |
| [68] | 1 PD line vs. 1 control line | V170G | iPSC-derived DA neurons | 1. Impairement in mitophagy |
| [67] | 7 PD lines vs. 5 control lines | Exon 4 or 7 deletion | iPSC-derived DA neurons | 1. Dysregulation of LRKK2 levels 2. Mitochondrial dysfunction |
2.5. iPSC Modelling of GBA Mutation and Associated Phenotypes
The GBA gene is located on chromosome 1 (1q21) and encodes for a lysosomal glucocerebrosidase enzyme (GCase), hydrolysing the glucosylceramide (GlcCer) into ceramide and glucose [74]. GBA mutation was first associated with PD approximately 14 years ago as a result of a PD like phenotype in PD with Gaucher disease [75]. The onset of PD with GBA mutations have been reported to be 30% at 80 years, with 9.1% of GBA carriers develop PD [76]. The main GBA mutations are p.N370S and p.L444P, enhancing the Lewy bodies formation, leading to PD and dementia [77]. Both mutations exhibit a reduced GCase activity that trigger an abnormal accumulation of α-synuclein [74]. Moreover, GBA has a key role in mitochondrial function and autophagy [78].
2.6. iPSC Modelling of DJ-1 Mutation and Associated Phenotypes
Dj-1 is a small protein with 189 amino acid residues, usually forming homodimers having a key role in anti-oxidant activities as well as directly inhibiting α-synuclein aggregation [79]. Mutations in DJ-1 have shown to cause early onset, autosomal recessive PD, either due to a base-pair deletion or a homozygous point mutation (L166P) [80][81]. The effects of mutations in DJ-1 on the development of PD has not been extensively studied in iPSC-derived DA neurons (Table 3). Increased dopamine oxidation and oxidative stress have been observed in iPSC-derived DA neurons, triggering mitochondrial oxidative stress, leading to the inactivation of glucocerebrosidaes [80]. This inactivation in turn inhibits lysosomal functions, elevating the level of α-synuclein, a known phenotype observed in iPSC- derived neurons from PD [80]. A more recent study utilized hiPSC-derived DA neurons carrying DJ-1 mutation and demonstrated a dysregulation in lysosomal proteins and activity [82][83][84].
Table 3. DJ-1-mutated iPSC-derived neuronal phenotype.
| Reference | Number of Cohorts | Type of Mutation | Cell Type | Phenotype |
|---|---|---|---|---|
| [83] | 1 PD isogonic lines vs. control lines | DJ-1 | iPSC-derived DA neurons | 1. Increased oxidative stress 2. Mitochondrial dysfunction 3. Lysosomal dysfunction |
| [80] | 3 PD lines vs. 2 control lines | c.192G>C | iPSC-derived DA neurons | 1. Increased oxidative stress 2. lysosomal dysfunction 3. α -synuclein aggregation |
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013.
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The emerging evidence of the parkinson pandemic. J. Parkinsons. Dis. 2018, 8, S3–S8.
- Lee, A.; Gilbert, R.M. Epidemiology of parkinson disease. Neurol. Clin. 2016, 34, 955–965.
- Savica, R.; Grossardt, B.R.; Bower, J.H.; Ahlskog, J.E.; Mielke, M.M.; Rocca, W.A. Incidence and time trends of drug-induced parkinsonism: A 30-year population-based study. Mov. Disord. 2017, 32, 227–234.
- Moisan, F.; Kab, S.; Mohamed, F.; Canonico, M.; Le Guern, M.; Quintin, C.; Carcaillon, L.; Nicolau, J.; Duport, N.; Singh-Manoux, A.; et al. Parkinson disease male-to-female ratios increase with age: French nationwide study and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 952–957.
- Ginis, P.; Nackaerts, E.; Nieuwboer, A.; Heremans, E. Cueing for people with Parkinson’s disease with freezing of gait: A narrative review of the state-of-the-art and novel perspectives. Ann Phys Rehabil Med 2018, 61, 407–413.
- Lim, I.; van Wegen, E.; de Goede, C.; Deutekom, M.; Nieuwboer, A.; Willems, A.; Jones, D.; Rochester, L.; Kwakkel, G. Effects of external rhythmical cueing on gait in patients with Parkinson’s disease: A systematic review. Clin. Rehabil. 2005, 19, 695–713.
- Gasser, T.; Hardy, J.; Mizuno, Y. Milestones in PD genetics. Mov. Disord. 2011, 26, 1042–1048.
- Pankratz, N.; Foroud, T. Genetics of Parkinson disease. Genet. Med. 2007, 9, 801–811.
- Ferreira, M.; Massano, J. An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol. Scand. 2017, 135, 273–284.
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Miraglia, F.; Ricci, A.; Rota, L.; Colla, E. Subcellular localization of alpha-synuclein aggregates and their interaction with membranes. Neural Regen. Res. 2018, 13, 1136–1144.
- Wersinger, C.; Sidhu, A. Attenuation of dopamine transporter activity by α-synuclein. Neurosci. Lett. 2003, 340, 189–192.
- Carnwath, T.; Mohammed, R.; Tsiang, D. The direct and indirect effects of α-synuclein on microtubule stability in the pathogenesis of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2018, 14, 1685–1695.
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48.
- Krüger, R.; Kuhn, W.; Leenders, K.L.; Sprengelmeyer, R.; Müller, T.; Woitalla, D.; Portman, A.T.; Maguire, R.P.; Veenma, L.; Schröder, U.; et al. Familial parkinsonism with synuclein pathology: Clinical and PET studies of A30P mutation carriers. Neurology 2001, 56, 1355–1362.
- Papapetropoulos, S.; Paschalis, C.; Athanassiadou, A.; Papadimitriou, A.; Ellul, J.; Polymeropoulos, M.H.; Papapetropoulos, T. Clinical phenotype in patients with alpha-synuclein Parkinson’s disease living in Greece in comparison with patients with sporadic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2001, 70, 662–665.
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5.
- Cooper, C.; Goldman, J.; Zabetian, C.; Mata, I.; Leverenz, J. SNCA G51D Missense Mutation Causing Juvenile Onset Parkinson’s Disease (P5. 8-026). Neurology 2019, 92.
- Li, X.; Tan, Y.-C.; Poulose, S.; Olanow, C.W.; Huang, X.-Y.; Yue, Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J. Neurochem. 2007, 103, 238–247.
- Roosen, D.A.; Cookson, M.R. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener. 2016, 11, 73.
- Gao, L.; Gómez-Garre, P.; Díaz-Corrales, F.J.; Carrillo, F.; Carballo, M.; Palomino, A.; Díaz-Martín, J.; Mejías, R.; Vime, P.J.; López-Barneo, J.; et al. Prevalence and clinical features of LRRK2 mutations in patients with Parkinson’s disease in southern Spain. Eur. J. Neurol. 2009, 16, 957–960.
- Alessi, D.R.; Sammler, E. LRRK2 kinase in Parkinson’s disease. Science 2018, 360, 36–37.
- Lee, J.-W.; Cannon, J.R. LRRK2 mutations and neurotoxicant susceptibility. Exp. Biol. Med. 2015, 240, 752–759.
- Di Fonzo, A.; Tassorelli, C.; De Mari, M.; Chien, H.F.; Ferreira, J.; Rohé, C.F.; Riboldazzi, G.; Antonini, A.; Albani, G.; Mauro, A.; et al. Italian Parkinson’s Genetics Network Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson’s disease. Eur. J. Hum. Genet. 2006, 14, 322–331.
- Marder, K.; Wang, Y.; Alcalay, R.N.; Mejia-Santana, H.; Tang, M.-X.; Lee, A.; Raymond, D.; Mirelman, A.; Saunders-Pullman, R.; Clark, L.; et al. LRRK2 Ashkenazi Jewish Consortium Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 2015, 85, 89–95.
- MacLeod, D.; Dowman, J.; Hammond, R.; Leete, T.; Inoue, K.; Abeliovich, A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 2006, 52, 587–593.
- Shin, N.; Jeong, H.; Kwon, J.; Heo, H.Y.; Kwon, J.J.; Yun, H.J.; Kim, C.-H.; Han, B.S.; Tong, Y.; Shen, J.; et al. LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 2008, 314, 2055–2065.
- Gómez-Suaga, P.; Rivero-Ríos, P.; Fdez, E.; Blanca Ramírez, M.; Ferrer, I.; Aiastui, A.; López De Munain, A.; Hilfiker, S. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum. Mol. Genet. 2014, 23, 6779–6796.
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schüle, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280.
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367.
- Shimura, H.; Hattori, N.; Kubo, S.I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305.
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608.
- Lücking, C.B.; Dürr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. French Parkinson’s Disease Genetics Study Group; European Consortium on Genetic Susceptibility in Parkinson’s Disease Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 2000, 342, 1560–1567.
- West, A.B.; Maidment, N.T. Genetics of parkin-linked disease. Hum. Genet. 2004, 114, 327–336.
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803.
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 2012, 5, 35.
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and PINK1 Patient iPSC-Derived Midbrain Dopamine Neurons Exhibit Mitochondrial Dysfunction and α-Synuclein Accumulation. Stem Cell Rep. 2016, 7, 664–677.
- Bogetofte, H.; Jensen, P.; Ryding, M.; Schmidt, S.I.; Okarmus, J.; Ritter, L.; Worm, C.S.; Hohnholt, M.C.; Azevedo, C.; Roybon, L.; et al. PARK2 Mutation Causes Metabolic Disturbances and Impaired Survival of Human iPSC-Derived Neurons. Front. Cell Neurosci. 2019, 13, 297.
- Aroso, M.; Ferreira, R.; Freitas, A.; Vitorino, R.; Gomez-Lazaro, M. New insights on the mitochondrial proteome plasticity in Parkinson’s disease. Proteomics Clin Appl 2016, 10, 416–429.
- Jin, J.; Hulette, C.; Wang, Y.; Zhang, T.; Pan, C.; Wadhwa, R.; Zhang, J. Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: Relevance to Parkinson disease. Mol. Cell Proteomics 2006, 5, 1193–1204.
- Hwang, O. Role of oxidative stress in Parkinson’s disease. Exp. Neurobiol. 2013, 22, 11–17.
- Jiang, H.; Ren, Y.; Zhao, J.; Feng, J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum. Mol. Genet. 2004, 13, 1745–1754.
- Jiang, H.; Jiang, Q.; Liu, W.; Feng, J. Parkin suppresses the expression of monoamine oxidases. J. Biol. Chem. 2006, 281, 8591–8599.
- Suzuki, S.; Akamatsu, W.; Kisa, F.; Sone, T.; Ishikawa, K.-I.; Kuzumaki, N.; Katayama, H.; Miyawaki, A.; Hattori, N.; Okano, H. Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochem. Biophys. Res. Commun. 2017, 483, 88–93.
- Chang, K.-H.; Lee-Chen, G.-J.; Wu, Y.-R.; Chen, Y.-J.; Lin, J.-L.; Li, M.; Chen, I.-C.; Lo, Y.-S.; Wu, H.-C.; Chen, C.-M. Impairment of proteasome and anti-oxidative pathways in the induced pluripotent stem cell model for sporadic Parkinson’s disease. Parkinsonism Relat. Disord. 2016, 24, 81–88.
- Jiang, H.; Ren, Y.; Yuen, E.Y.; Zhong, P.; Ghaedi, M.; Hu, Z.; Azabdaftari, G.; Nakaso, K.; Yan, Z.; Feng, J. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat. Commun. 2012, 3, 668.
- Zhong, P.; Hu, Z.; Jiang, H.; Yan, Z.; Feng, J. Dopamine Induces Oscillatory Activities in Human Midbrain Neurons with Parkin Mutations. Cell Rep. 2017, 19, 1033–1044.
- Shaltouki, A.; Sivapatham, R.; Pei, Y.; Gerencser, A.A.; Momčilović, O.; Rao, M.S.; Zeng, X. Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem Cell Rep. 2015, 4, 847–859.
- Konovalova, E.V.; Lopacheva, O.M.; Grivennikov, I.A.; Lebedeva, O.S.; Dashinimaev, E.B.; Khaspekov, L.G.; Fedotova, E.Y.; Illarioshkin, S.N. Mutations in the Parkinson’s Disease-Associated PARK2 Gene Are Accompanied by Imbalance in Programmed Cell Death Systems. Acta Naturae 2015, 7, 146–149.
- Shiba-Fukushima, K.; Ishikawa, K.-I.; Inoshita, T.; Izawa, N.; Takanashi, M.; Sato, S.; Onodera, O.; Akamatsu, W.; Okano, H.; Imai, Y.; et al. Evidence that phosphorylated ubiquitin signaling is involved in the etiology of Parkinson’s disease. Hum. Mol. Genet. 2017, 26, 3172–3185.
- Okarmus, J.; Bogetofte, H.; Schmidt, S.I.; Ryding, M.; García-López, S.; Ryan, B.J.; Martínez-Serrano, A.; Hyttel, P.; Meyer, M. Lysosomal perturbations in human dopaminergic neurons derived from induced pluripotent stem cells with PARK2 mutation. Sci. Rep. 2020, 10, 10278.
- Zanon, A.; Kalvakuri, S.; Rakovic, A.; Foco, L.; Guida, M.; Schwienbacher, C.; Serafin, A.; Rudolph, F.; Trilck, M.; Grünewald, A.; et al. SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin-deficient human iPSC-derived neurons and Drosophila. Hum. Mol. Genet. 2017, 26, 2412–2425.
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131.
- Albanese, A.; Valente, E.M.; Romito, L.M.; Bellacchio, E.; Elia, A.E.; Dallapiccola, B. The PINK1 phenotype can be indistinguishable from idiopathic Parkinson disease. Neurology 2005, 64, 1958–1960.
- Ibáñez, P.; Lesage, S.; Lohmann, E.; Thobois, S.; De Michele, G.; Borg, M.; Agid, Y.; Dürr, A.; Brice, A. French Parkinson’s Disease Genetics Study Group Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain 2006, 129, 686–694.
- Bonifati, V.; Rohé, C.F.; Breedveld, G.J.; Fabrizio, E.; De Mari, M.; Tassorelli, C.; Tavella, A.; Marconi, R.; Nicholl, D.J.; Chien, H.F.; et al. Italian Parkinson Genetics Network Early-onset parkinsonism associated with PINK1 mutations: Frequency, genotypes, and phenotypes. Neurology 2005, 65, 87–95.
- Lin, W.; Kang, U.J. Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 2008, 106, 464–474.
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769.
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160.
- Arena, G.; Valente, E.M. PINK1 in the limelight: Multiple functions of an eclectic protein in human health and disease. J. Pathol. 2017, 241, 251–263.
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91.
- Seibler, P.; Graziotto, J.; Jeong, H.; Simunovic, F.; Klein, C.; Krainc, D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 2011, 31, 5970–5976.
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153.
- Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20.
- Azkona, G.; López de Maturana, R.; Del Rio, P.; Sousa, A.; Vazquez, N.; Zubiarrain, A.; Jimenez-Blasco, D.; Bolaños, J.P.; Morales, B.; Auburger, G.; et al. LRRK2 Expression Is Deregulated in Fibroblasts and Neurons from Parkinson Patients with Mutations in PINK1. Mol. Neurobiol. 2018, 55, 506–516.
- Rakovic, A.; Shurkewitsch, K.; Seibler, P.; Grünewald, A.; Zanon, A.; Hagenah, J.; Krainc, D.; Klein, C. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: Study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem. 2013, 288, 2223–2237.
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117.
- Bogaerts, V.; Theuns, J.; van Broeckhoven, C. Genetic findings in Parkinson’s disease and translation into treatment: A leading role for mitochondria? Genes Brain Behav. 2008, 7, 129–151.
- Hoepken, H.-H.; Gispert, S.; Morales, B.; Wingerter, O.; Del Turco, D.; Mülsch, A.; Nussbaum, R.L.; Müller, K.; Dröse, S.; Brandt, U.; et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis. 2007, 25, 401–411.
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.Y.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, I.; Klupsch, K.; Deas, E.; et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 2008, 3, e2455.
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra90.
- Straniero, L.; Rimoldi, V.; Samarani, M.; Goldwurm, S.; Di Fonzo, A.; Krüger, R.; Deleidi, M.; Aureli, M.; Soldà, G.; Duga, S.; et al. The GBAP1 pseudogene acts as a ceRNA for the glucocerebrosidase gene GBA by sponging miR-22-3p. Sci. Rep. 2017, 7, 12702.
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36.
- Anheim, M.; Elbaz, A.; Lesage, S.; Durr, A.; Condroyer, C.; Viallet, F.; Pollak, P.; Bonaïti, B.; Bonaïti-Pellié, C.; Brice, A. French Parkinson Disease Genetic Group Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012, 78, 417–420.
- Velayati, A.; Yu, W.H.; Sidransky, E. The role of glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr Neurol Neurosci Rep 2010, 10, 190–198.
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356.
- Wilson, M.A. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid. Redox Signal. 2011, 15, 111–122.
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261.
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259.
- Hsieh, C.-H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic parkinson’s disease. Cell Stem Cell 2016, 19, 709–724.
- Ahfeldt, T.; Ordureau, A.; Bell, C.; Sarrafha, L.; Sun, C.; Piccinotti, S.; Grass, T.; Parfitt, G.M.; Paulo, J.A.; Yanagawa, F.; et al. Pathogenic Pathways in Early-Onset Autosomal Recessive Parkinson’s Disease Discovered Using Isogenic Human Dopaminergic Neurons. Stem Cell Rep. 2020, 14, 75–90.
- Xu, C.-Y.; Kang, W.-Y.; Chen, Y.-M.; Jiang, T.-F.; Zhang, J.; Zhang, L.-N.; Ding, J.-Q.; Liu, J.; Chen, S.-D. DJ-1 Inhibits α-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front. Aging Neurosci. 2017, 9, 308.
More
Information
Subjects:
Neurosciences
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
07 Jul 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No