Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Catherine YZYDORCZYK | + 4230 word(s) | 4230 | 2021-07-02 11:47:28 | | | |
| 2 | Camila Xu | Meta information modification | 4230 | 2021-07-06 09:48:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yzydorczyk, C.; Peyter, A. Metabolic Syndrome (MetS). Encyclopedia. Available online: https://encyclopedia.pub/entry/11704 (accessed on 07 February 2026).
Yzydorczyk C, Peyter A. Metabolic Syndrome (MetS). Encyclopedia. Available at: https://encyclopedia.pub/entry/11704. Accessed February 07, 2026.
Yzydorczyk, Catherine, Anne-Christine Peyter. "Metabolic Syndrome (MetS)" Encyclopedia, https://encyclopedia.pub/entry/11704 (accessed February 07, 2026).
Yzydorczyk, C., & Peyter, A. (2021, July 06). Metabolic Syndrome (MetS). In Encyclopedia. https://encyclopedia.pub/entry/11704
Yzydorczyk, Catherine and Anne-Christine Peyter. "Metabolic Syndrome (MetS)." Encyclopedia. Web. 06 July, 2021.
Copy Citation
Metabolic syndrome (MetS) is a cluster of several disorders, such as hypertension, central obesity, dyslipidemia, hyperglycemia, insulin resistance and non-alcoholic fatty liver disease.
developmental programming
intrauterine growth restriction
metabolic syndrome
endothelial progenitor cells
oxidative stress
cellular senescence
1. Components of Metabolic Syndrome
The incidence and prevalence of metabolic syndrome (MetS) are increasing worldwide and MetS is becoming a global health problem. MetS affects 20–30% of the population in developed countries [1]. The major components of the MetS cluster are obesity, in particular abdominal body fat accumulation, impaired glucose metabolism, dyslipidemia and arterial hypertension [2][3]. Additionally, non-alcoholic fatty liver disease (NAFLD), which is defined as excess fat (>5% weight or volume) deposition in the liver in the absence of excessive alcohol intake, has been identified as a hepatic manifestation of MetS [4]. NAFLD has emerged as the most common cause of liver disease, and it is associated with substantial morbidity and mortality in Western countries. Moreover, MetS has been identified as an epidemiologic tool related to cardiovascular disease (CVD) risk. In fact, each component of the MetS represents an independent risk factor for the development of CVD [5][6][7].
2. Metabolic Syndrome and Endothelial Dysfunction
The endothelium is a thin monocellular layer that covers the inner surface of blood vessels, separating the circulating blood from the interstitial fluid [8], and plays an essential role in the maintenance of vascular homeostasis [9]. Under physiological conditions, the endothelium synthesizes paracrine factors, such as nitric oxide (NO), prostacyclin, endothelium-derived hyperpolarizing factor and natriuretic peptide type-C. These factors regulate the balance between vasodilation and vasoconstriction, inhibit and improve the proliferation and migration of smooth muscle cells, prevent and stimulate the adhesion and aggregation of platelets, and regulate thrombogenesis and fibrinolysis [10][11][12]. NO is the major contributor of these endothelial functions. NO is a gaseous molecule generated during the conversion of L-arginine to L-citrulline via the action of the endothelial nitric oxide synthase (eNOS), which requires the presence of tetrahydrobiopterin (BH4) as cofactor [13]. Endothelial function can be explored notably by infusion of acetylcholine, which stimulates endothelial muscarinic receptors, leading to an increase in cytosolic Ca2+ and thus to the activation of eNOS to metabolize L-arginine into L-citrulline and NO [14]. Measurement of flow-mediated vasodilation (FMD) by ultrasound allows a noninvasive assessment of endothelial function in patients [15]. Given the large range of the vasoprotective effects of NO, the term “endothelial dysfunction” generally refers to reduced NO bioavailability, notably due to decreased eNOS expression or activity, resulting in enhanced vasoconstrictor responses and thus impaired endothelium-dependent vasodilation [16].
Therefore, an alteration in the structural and functional integrity of the endothelium can lead to the development of cardiometabolic disorders. All components of MetS can individually be associated with impaired endothelial function. Several studies have shown that hypertension [17][18] and abdominal obesity [19][20][21] are associated with endothelial dysfunction. In patients with type-2 diabetes, elevated glucose levels lead to glycosylation of vascular endothelium, resulting in changes in blood vessels such as narrowing and sprouting of neovasculature that is friable and at risk of rupture [22][23][24]. It has been shown that patients with type-2 diabetes have a 2- to 4-fold increased risk of developing CVD compared with non-diabetic individuals [25], related to endothelial dysfunction, as a consequence of inflammation, increased reactive oxygen species (ROS) production and deletion of eNOS [26][27][28]. Patients with NAFLD and non-alcoholic steatohepatitis also display an impaired endothelial function characterized by decreased FMD [29][30][31].
3. Endothelial Progenitor Cells
The endothelial progenitor cells (EPCs) are circulating components of the endothelium. They are mobilized and migrate in the circulation from the bone marrow, differentiate into mature endothelial cells, and synthesize and release a wide range of active molecules and growth factors modulating vasculogenesis and improving vascular homeostasis [32][33]. A close association has been identified between the maintenance of endothelial structure and function by EPCs and their ability to differentiate and repair damaged endothelial tissue [34]. EPCs are most frequently isolated from cord blood [35][36] or peripheral blood [35][37], but can also be isolated from pulmonary artery endothelium [38][39] or placenta [40], or can be derived from induced pluripotent stem cells [41]. EPCs can be distinguished according to their phenotype and functional properties in vivo [42]. Early EPCs have a hematopoietic origin and promote angiogenesis through paracrine mechanisms but cannot give rise to mature endothelial cells [42][43][44][45]. In contrast, endothelial colony forming cells (ECFCs) or late outgrowth EPCs [42] have clonal potential and the capabilities to yield mature endothelial cells and to promote vascular formation in vitro and in vivo. In particular, these cells are capable of proliferation, autorenewal, migration, differentiation, vascular growth and neovascularization. It has been demonstrated that ECFCs can be characterized by the assessment of surface markers, such as CD34 and vascular endothelial growth factor receptor-2 (VEGFR-2, also named KDR) [46][47], but the absence of CD45. Importantly, the CD34+KDR+ combination is the only putative ECFCs phenotype that has been repeatedly and convincingly demonstrated to be an independent predictor of cardiovascular outcomes [48][49]. EPCs have not only a potential therapeutic impact on endothelial dysfunction in both clinical [50] and experimental [51] studies, but also the circulating levels of EPCs can be used as a clinical marker of disease progression [52]. EPCs dysfunction has been characterized by decreased number and/or impaired function of circulating precursors [53]. The number of circulating EPCs was found to be negatively correlated with cardiovascular risk factors and vascular function and to predict CVD independently of both conventional and non-traditional cardiovascular risk factors [54][55][56].
4. Endothelial Progenitor Cells Dysfunction in Cardiometabolic Disorders
4.1. Endothelial Progenitor Cells Dysfunction in Type-2 Diabetes
In patients with type-2 diabetes, a decrease in the number of EPCs has been reported, and the number of EPCs was lower as more numerous were the complications [57]. Notably a reduction in CD34+ EPCs has been mentioned in early stages of type-2 diabetes, which persists thereafter and worsens in patients with diabetes complications [58]. EPCs isolated from patients with type-2 diabetes have demonstrated impaired functions such as alterations of proliferation, migration, chemokinesis, angiogenesis and NO bioavailability [59] compared to nondiabetic patients [60][61][62], as observed also in EPCs from animal models of diabetes [63][64][65][66].
In addition, it is well known that endothelial dysfunction induced by hyperglycemia leads to micro- and macro-angiopathies complications [67]. A reduced number of EPCs in type-2 diabetes has been associated to increased brachial-ankle pulse wave velocity related to arterial stiffness [68] and is correlated to the prevalence of peripheral vascular disease [69][70][71] and with the degree of atherosclerosis [72]. Additionally, high glucose might impair EPCs by modifying NO-related mechanisms [73].
4.2. Endothelial Progenitor Cells Dysfunction in Hypertension and Cardiovascular Diseases
EPCs seem to play a protective role against the development of CVD [74]. In fact, several CVD have been associated with altered EPCs number and functions. In prehypertensive patients, an impaired formation of EPCs colonies has been mentioned [75]. Patients with hypertension and vascular lesions displayed a reduced number of circulating CD34+ cells [76]. In contrast, Skrzypkowska et al. observed that patients with essential hypertension had increased proportions of CD34+ EPCs [77], which may be a compensatory mechanism. Marketou et al. did not find any significant difference in the number of circulating CD34+ cells between hypertensive and normotensive individuals, but found a correlation between the number of circulating CD34+ cells and pulse wave velocity in hypertensive patients, suggesting a role for EPCs in the pathophysiology of arterial stiffness and arterial remodeling [78]. Hypertension is a risk factor for the incidence of other CVD such as stroke, coronary artery disease, sudden death, heart failure and peripheral arterial disease [79][80]. Hill et al. found a correlation between the number of circulating EPCs and the patient’s combined Framingham risk factor score, which includes six coronary risk factors, such as age, gender, total cholesterol, high density lipoprotein cholesterol, smoking habits and systolic blood pressure value [81][82]. Vasa et al., showed an impaired migration function of EPCs in patients with coronary artery disease [83]. In hypertensive patients with left ventricular hypertrophy, the circulating levels and adhesive function of EPCs were lowered compared to non-hypertensive patients [84]. A significant reduction of EPCs number and proliferation rate has been also observed in patients with peripheral artery disease [85] alone and combined with diabetes [70][86].
4.3. Endothelial Progenitor Cells Dysfunction in Obesity
In obese patients, a decrease in EPCs number associated with significantly impaired clonogenic properties and an altered capacity to incorporate into tubule structures has been observed [87]; the decrease in EPCs number was reversed by weight loss [88][89]. In a C57BL/6J mice model of obesity induced by high fat diet, the number of EPCs from adipose tissue was significantly lower, as well as the circulating level of EPCs in response to ischemia, compared to control mice. The colony-forming capacity of peripheral blood-derived EPCs and the angiogenic capacity in response to ischemic stimulation were markedly altered in the obese mice compared to controls [90]. In addition, an impaired recovery of damaged endothelium, reduced EPCs angiogenesis ability and left ventricular ejection fraction, and an increased left ventricular remodeling have been observed in the obese compared to control mice [91].
4.4. Endothelial Progenitor Cells Dysfunction and Dyslipidemia
Dyslipidemia is characterized by increased triglyceride concentrations, decreased plasma high-density lipoprotein (HDL)-cholesterol levels and an increased proportion of small, dense low-density lipoprotein (LDL) particles, despite normal LDL-cholesterol. Hypercholesterolemia has been associated with reduced EPCs availability [92][93]. In vitro exposure of EPCs to oxidized-LDL decreased their number and impaired their adhesive, migratory and tube-formation capacities in a dose-dependent manner [94].
4.5. Endothelial Progenitor Cells Dysfunction and NAFLD
NAFLD is currently well recognized as a hepatic manifestation of MetS [95] and has been associated with obesity, insulin resistance, systemic inflammation and advanced atherosclerosis [96][97]. In addition, NAFLD has been related to endothelial dysfunction [98]. Patients with NAFLD have decreased number and function of circulating EPCs associated with features of MetS [99]. However, it has been shown that the number of EPCs was higher in patients with NAFLD and MetS in comparison to those without these conditions, and that the EPCs number was directly proportional to the degree of liver steatosis. The increase in EPCs number could be considered as a compensatory mechanism against endothelial injury [100].
5. Mechanisms Potentially Associated with Impaired Functionality of Endothelial Progenitor Cells
Several mechanisms have been identified to impair EPCs functionality (Figure 1).
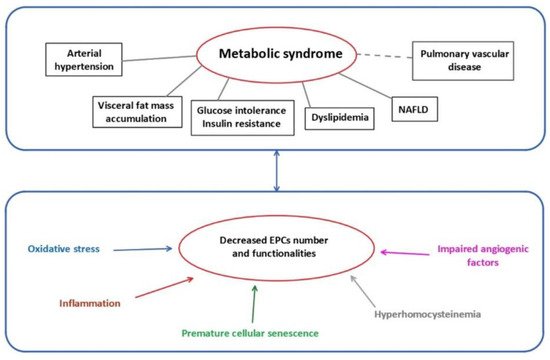
Figure 1. Metabolic syndrome and mechanisms related to EPCs dysfunctions. EPCs: endothelial progenitor cells; NAFLD: non-alcoholic fatty liver disease.
5.1. Oxidative Stress
ROS are chemically reactive molecules formed during the metabolism of molecular oxygen. ROS are necessary in several biochemical processes such as intracellular signaling, cell differentiation, growth arrest, apoptosis, immunity and defense against microorganisms. The natural antioxidant system consists of a series of antioxidant enzymes, such as superoxide dismutases (SOD), catalase and glutathione peroxidase, and of endogenous antioxidant compounds. Oxidative stress occurs when the amount of ROS exceeds the antioxidant capacity. Excessive ROS production can interact with cellular macromolecules and then enhance the process of lipid peroxidation, cause DNA damage and/or induce protein and nucleic acid modifications [101], leading to decreased biological activity, dysregulated metabolism and alterations in cell signaling. In addition, oxidative stress can affect NO synthesis and bioavailability. Superoxide anion is known to interact with NO, leading to the formation of peroxynitrite, a highly reactive and toxic species able to modify macromolecules, such as lipids, proteins and DNA. Moreover, it has been suggested that increased oxidation of BH4 to 7,8-dihydropteridine (BH2) results in a reduced availability of this cofactor for eNOS [102], which impairs its activity and therefore NO production. Additionally, oxidative stress leads to increased endothelial-derived constricting factors such as endothelin-1 (ET-1), angiotensin II, thromboxane A2 and prostaglandin H2, thus enhancing vasoconstriction and contributing to endothelial dysfunction [103]. In addition, asymmetric dimethylarginine (ADMA), an endogenous competitive inhibitor of eNOS, inhibits the formation of NO and can lead, via eNOS uncoupling, to increased superoxide radical generation. Oxidative stress has been associated with decreased EPCs levels associated with reduced capability of mobilizing, migrating and incorporating into existing vasculature [62][104][105][106].
The expression of antioxidant enzymes, such as SOD, catalase and glutathione peroxidase, in EPCs is higher compared to endothelial cells [107] in the purpose to improve EPCs survival within the oxygen-poor environment of the bone marrow, as well as to support the ability of EPCs to engraft within ischemic tissues during the vasculogenesis process [107][108][109][110]. Dyslipidemia can lead to oxidative stress and so to EPCs dysfunction. In fact, oxidized-LDL, as oxygen donors, are responsible for inciting and perpetuating oxidative stress through a spiral of redox-based reactions, which impairs the vasculogenic function of EPCs [111]. In contrast, HDL, which have antioxidant and anti-inflammatory properties, have a positive impact on EPCs physiology [112].
Mitochondria play an important role in energy homeostasis by the production of ATP via oxidative phosphorylation and the oxidation of metabolites via the Krebs’s cycle and the beta-oxidation of fatty acids. Mitochondrial dysfunction is the major source of ROS production (0.2% to 2% of total oxygen taken up by cells), mainly at complex I (NADH CoQ reductase) and complex III (bc1 complex). Under normal conditions, the overproduction of ROS in mitochondria is restricted via enzymatic and non-enzymatic defense systems to protect cellular organelles from oxidative damage. However, when antioxidant defenses are overwhelmed, there is an overproduction of ROS, which then leads to oxidative damage to proteins, DNA and lipids in mitochondria [113][114]. An altered mitochondrial activity in EPCs has been observed in patients with cerebrovascular disorder [115] and type-2 diabetes [116].
5.2. Cellular Senescence
Cellular senescence is a biological phenomenon triggered by potentially harmful stimuli, during which the cell interrupts the division process, entering a state of cell cycle arrest and becoming quiescent. Senescence is a protective mechanism affecting the majority of the cells within an organism [117]. At the phenotypic level, senescent cells acquire a characteristic flattened and enlarged morphology with accumulation of lipofuscin, which is a marker of highly oxidized, insoluble proteins [118]. Cellular senescence is also characterized by a decline in the DNA replication in the cells, until they cease to proliferate, associated with molecular changes in elements related to the cell cycle (pRb, p21WAF, p16INK4a and p53) [119]. Moreover, senescent cells undergo chromatin and secretome changes, genomic and epigenomic damage, unbalanced mitogenic signals and tumor-suppressor activation [120]. In addition to these common characteristics, replicative senescence can be identified by a decline in telomere length with each cell cycle [121]. Replicative senescence is an irreversible phenomenon, in contrast to stress-induced premature senescence (SIPS). SIPS is initiated in young cells via different mechanisms, such as oxidative stress, and has been associated with the over-expression of p16INK4a and the decreased functionality of the anti-aging protein sirtuin-1 [122]. Moreover, senescent cells can exhibit an upregulation and secretion of growth factors, such as proinflammatory cytokines (IL-6 and IL-1), chemokines (IL-8, chemokine ligands family members), macrophage inflammatory protein and insulin-like growth factor, and also a release of extracellular matrix-degrading proteins (MMP family, serine proteases and fibronectin); the overall effect of these phenomena leads to the senescence-associated secretory phenotype (SASP) [123]. It is important to note that most senescent cells are resistant to some apoptosis signals, therefore they become senescent [119].
EPCs isolated from cord blood of diabetic mothers displayed in vitro premature senescence and impaired proliferation and in vivo reduced vasculogenic potential compared to uncomplicated pregnancies [124]. Oxidative stress and senescence are hallmarks often associated. An increased ROS production as well as oxidized-LDL have been associated with cellular senescence of EPCs [109][125], decreasing their number and impairing their function. Moreover, an association has been observed between oxidative DNA damage, decreased telomerase activity and decreased telomere length in EPCs isolated from patients with MetS and coronary artery disease [126].
5.3. Impaired Angiogenic Function
Angiogenesis is important to maintain the integrity of tissue perfusion, which is crucial for physiologic organ function. EPCs, and more particularly ECFCs, play a major role in the angiogenic process. In fact, it has been shown that ECFCs contribute not only to maintain microvasculature but also to stimulate postnatal angiogenesis [127][128][129].
NO is necessary for angiogenesis to occur [130]. NO is involved in the mobilization of EPCs and improves their migratory and proliferative activities [131], notably by the regulation of their angiogenic activity [132][133]. NO-mediated signaling pathways are essential for EPCs mobilization from the bone marrow [134][135][136]. NO regulates migration of EPCs into ischemic sites [61][136][137] and improves their survival [138]. A link between eNOS expression/functionality and EPCs function has been described [139]. In diabetic EPCs, eNOS activity is decreased, probably due to eNOS uncoupling, leading to reduced NO production and thus to decreased migratory capability, which is restored by exogenous NO administration [136][140][141]. The NO-donor sodium nitroprusside improved migration and tube formation, which was impaired by hyperglycemia [142]. ADMA levels are increased in diabetes [143] and it has been shown that ADMA decreased EPCs proliferation and differentiation, in a concentration-dependent manner [106].
NO can interact with angiogenic factors. Vascular endothelial growth factor (VEGF) plays an important role in EPCs differentiation and vascular repair [144][145]. A reciprocal relation between NO and VEGF has been demonstrated. The synthesis of VEGF can be induced by NO [146][147], and VEGF increases NO production by eNOS, promoting angiogenesis [148]. In patients with coronary heart disease, a reduced NO bioavailability has been observed, associated with altered VEGF expression and subsequently impaired EPCs functionality [149][150]. Moreover, a decreased secretion of NO and VEGF induced by hyperglycemia condition or advanced glycation end-products decreased activity of SOD, and so impaired EPCs function, such as migration and tube formation [151].
5.4. Inflammation-Induced EPCs Dysfunction
EPCs function and maturation are extremely sensitive to inflammation mediators. Autoimmune diseases like systemic lupus erythematosus (SLE) or rheumatoid arthritis as well as metabolic anomalies like type-2 diabetes mellitus demonstrate endothelial dysfunction related to chronic inflammation and later to CVD [152]. Patients with SLE displayed a chronic inflammatory state and a higher risk of MetS associated with a decreased level of circulating EPCs [153]. Interestingly, adipokines are critical mediators of inflammation and insulin resistance in SLE-associated MetS [154].
The level of type-I interferon impacts the EPCs number and function [155], especially in reducing their ability to repair vascular damage [156][157]. In a murine model, a type-1 interferon receptor knockout led to increased EPCs number and function, with improved neoangiogenesis and cell differentiation [158]. The blockade of IL-18 also enhanced differentiation of EPCs [159]. Increased expression of tumor necrosis factor α (TNFα) had detrimental effects on EPCs function as impaired proliferation, migration and tube formation [160]. Stromal cell-derived factor-1 (SDF-1) is a cytokine stimulating the recruitment of proinflammatory cells that contributes to EPCs mobilization [161]. In patients with type-2 diabetes, hyperglycemia reduces the level of VEGF and SDF-1 secretion from endothelial cells via the hypoxia-inducible factor/hypoxia-response element pathway and dipeptidyl peptidase-4 activity [162], therefore decreasing the mobilization of EPCs from bone marrow to circulation [163][164] and impairing the regulation of growth, migration and survival of EPCs [165].
5.5. Epigenetic Regulation
5.5.1. MicroRNAs
MicroRNAs (miRNAs) have been identified to play important roles in the post-transcriptional regulation of gene expression influencing several cellular processes which contribute to disease [166][167]. Several miRNAs have been identified to regulate endothelial cell functions, such as cell proliferation, senescence, migration, differentiation and vascular tubule formation [168][169][170]. Therefore, an alteration of miRNAs expression can contribute to EPCs dysfunctions. It has been shown that miR-126 expression is necessary to downregulate Spred-1 to activate Ras/ERK/VEGF and PI3K/AKT/eNOS signaling pathways, improving EPCs proliferation and migration [171]. MiR-130a expression is involved in the proliferation, migration and colonies formation via RUNX3/ ERK/VEGF and PI3K/AKT signaling pathways [172]. Additionally, miR-31 is involved in the expression of several proteins implicated notably in the differentiation of bone-forming stem cells into mesenchymal and fat tissues [173][174]. In EPCs isolated from patients with type-2 diabetes, a downregulation of miR-126 and miR-130a, but an increase in miR-31 expression have been associated with an impairment of their function [171][172][175]. In addition, miR-34a overexpression led to increased EPCs senescence, associated with decreased sirtuin-1 functionality [176] and elevated miR-31-triggered apoptosis, which impaired EPCs functions in diabetic patients [177].
5.5.2. DNA Methylation
DNA methylation is the best-known epigenetic mechanism, and usually leads to repressed transcription of the involved gene. Methylation takes place on CpG islands located mainly in the promoter region of the genes. Usually, if the CpG islands in the promoter region are unmethylated, the gene is transcribed, but when a significant part of these islands is methylated, the gene can no longer be transcribed, being so silenced. DNA methylation can be altered by early environmental factors [178]. Methyl CpG binding protein 2 (MeCP2) is an important member of the methyl-CpG binding protein family. It has been shown that overexpression of MeCP2 reduced angiogenesis, via decreased protein levels of p-eNOS/eNOS and VEGF and induced senescent EPCs dysfunction through sirtuin-1 promoter hypermethylation [179].
5.5.3. Histone Modification
In the nucleus, DNA is packaged into chromatin as repeating units of nucleosomes, which form a “beads-on-a-string” structure that can compact into higher order structures to affect gene expression. Nucleosomes are composed of 146-bp DNA wrapped in histone octamers (composed of two H2A, H2B, H3 and H4) and are connected by a linker DNA, which can associate with histone H1 to form heterochromatin. Histone proteins contain a globular domain and an amino-terminal tail, with the latter being post-translationally modified. The post-translational modifications of lysine (acetylation, methylation, ubiquitination, sumoylation), arginine (methylation), as well as serine and threonine (phosphorylation) are the most described. It has been shown that the transcriptionally active H3K4me3 state leads to the activation of multiple pro-angiogenic signaling pathways (VEGFR, CXCR4, WNT, NOTCH, SHH) which improved the capacity of EPCs to form capillary-like networks in vitro and in vivo [180]. The concomitant inhibition of silencing histone modification (H3K27me3) and enhancement of activating histone modification (H3K4me3) improved eNOS expression in EPCs [181].
5.6. Hyperhomocysteinemia
Hyperhomocysteinemia is a clinical condition characterized by a high level of homocysteine in blood (above 15 µmol/L) [182][183][184]. Homocysteine is a sulfur-containing amino acid synthesized during the metabolism of methionine. It is catabolized either by remethylation to methionine, catalyzed by methionine synthase or betaine homocysteine methyl transferase, or by transsulfuration, catalyzed by cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE), leading to cysteine, a precursor of glutathione [185][186][187]. CBS and CSE require vitamin B6 as co-factor. A rate-limiting CBS enzyme as well as an insufficient dietary supply of cofactors have been involved in severe cases of hyperhomocysteinemia [188][189][190]. Other causes have also been identified, such as smoking, renal failure and other systemic diseases [191].
Hyperhomocysteinemia is an independent risk factor for cardiovascular disorders [192]. It has been associated with endothelial dysfunction in several animal models [193][194][195]. Hyperhomocysteinemia has been shown to contribute to endothelial dysfunction by induction of oxidative stress. In fact, hyperhomocysteinemia increases inducible nitric oxide synthase (iNOS) synthesis and ROS production [196], associated with SOD inactivation [197][198], so generating important quantities of peroxynitrite [196][199] and, therefore, contributing to nitrative stress which induces severe damage to proteins, lipids and DNA [200][201][202]. EPCs are particularly sensitive to oxidative stress, so hyperhomocysteinemia can contribute to EPCs dysfunction. It has been shown that homocysteine dose- and time-dependently impaired EPCs proliferative, migratory, adhesive and in vitro vasculogenesis capacities [203]. A significant decrease in circulating EPCs number and impaired functional capacity were observed in patients with hyperhomocysteinemia [204]. In mice, hyperhomocysteinemia-induced nitrative stress contributed directly to the injury of EPCs, by decreasing their survival rate, inducing apoptosis and necrosis [205]. In patients with stroke, high plasma level of homocysteine has been associated with a reduced number of EPCs colonies related to apoptosis [206][207], and administration of B vitamins (B6, B9) was able to attenuate such effects [206]. DNA methylation is an important process for gene transcription and therefore for regulation of protein expression. EPCs isolated from bone marrow in a mice model fed with a high methionine-rich diet displayed a reduced adhesion capacity and tube formation abilities associated with hyper-methylation in the CpG islands of the CBS promoter, leading to downregulated CBS expression. Such dysfunctions were reversed by administration of a DNA methylation inhibitor to mice fed with a high methionine-rich diet [208].
Homocysteine and hydrogen sulfide (H2S) are interconnected. H2S is an endogenous gasotransmitter, produced by CSE, CBS and 3-mercaptopyruvate sulfur transferase [187][209][210]. H2S acts via the S-sulfhydration of cysteine residues (-SH) of target proteins to form persulfide group (-SSH), which can modify the structure and activity of various target proteins [210][211]. H2S is emerging as an essential contributor to homeostasis of endothelial function, besides NO. It is involved in the regulation of several systems such as cardiovascular, nervous, gastrointestinal and renal systems, but also in the inflammatory and immune responses [212]. In the cardiovascular system, H2S is produced in cardiomyocytes, vascular endothelial cells, smooth muscle cells and EPCs [213]. H2S exerts antioxidant, anti-apoptotic, anti-inflammatory and vasoactive activities, and regulates proliferation, migration and angiogenesis, in an autocrine and paracrine manner [209][210][214][215][216]. H2S induces vasorelaxation by opening of KATP channels in vascular smooth muscle cells and partially through a K+ conductance in endothelial cells [217]. H2S can decrease inflammation by inhibiting transcription factors such as NF-kB [218]. H2S can reduce oxidative stress, through direct scavenging of oxygen and nitrogen species and enhancing antioxidant defense mechanisms, notably via the Keap1/Nrf2 pathway, and delays senescence [219]. In addition, H2S can also interact with the NO/NOS pathway to control vascular function, thanks the inhibition of phosphodiesterases in smooth muscle cells, by PI3K/AKT-dependent phosphorylation of eNOS in Ser1177 and by stabilization of eNOS in the dimeric state [220][221][222]. Moreover, H2S is a major factor to ensure EPCs functionality. In diabetic leptin receptor deficient db/db mice, the H2S plasma levels were significantly reduced and associated with impaired EPCs functionality such as tube formation, adhesive function and wound healing by decreasing angiogenesis process [223]. Shear stress was found to improve several EPCs functions, such as proliferation, migration, tube formation and reendothelialization [224][225][226], probably through enhancement of H2S production [227]. Indeed, shear stress was able to increase H2S production and CSE protein expression in human EPCs in a dose- and time-dependent manner, and to improve EPCs proliferation, migration and adhesion capacity [228].
As biogenesis of H2S and homocysteine is regulated by each other and imbalance between both molecules seems implicated in several cardiovascular disorders, the H2S/homocysteine ratio could be useful for cardiovascular risk prediction [186].
References
- Spahis, S.; Borys, J.M.; Levy, E. Metabolic Syndrome as a Multifaceted Risk Factor for Oxidative Stress. Antioxid. Redox. Signal. 2017, 26, 445–461.
- Bonomini, F.; Rodella, L.F.; Rezzani, R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015, 6, 109–120.
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C., Jr.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005, 112, 2735–2752.
- Stefan, N.; Haring, H.U.; Cusi, K. Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324.
- Sperling, L.S.; Mechanick, J.I.; Neeland, I.J.; Herrick, C.J.; Despres, J.P.; Ndumele, C.E.; Vijayaraghavan, K.; Handelsman, Y.; Puckrein, G.A.; Araneta, M.R.; et al. The CardioMetabolic Health Alliance: Working Toward a New Care Model for the Metabolic Syndrome. J. Am. Coll. Cardiol. 2015, 66, 1050–1067.
- Ritchie, S.A.; Connell, J.M. The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 319–326.
- Berwick, Z.C.; Dick, G.M.; Tune, J.D. Heart of the matter: Coronary dysfunction in metabolic syndrome. J. Mol. Cell. Cardiol. 2012, 52, 848–856.
- Luscher, T.F.; Barton, M. Biology of the endothelium. Clin. Cardiol. 1997, 20, II-3–10.
- Martin, F.A.; Murphy, R.P.; Cummins, P.M. Thrombomodulin and the vascular endothelium: Insights into functional, regulatory, and therapeutic aspects. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1585-97.
- Minshall, R.D.; Tiruppathi, C.; Vogel, S.M.; Malik, A.B. Vesicle formation and trafficking in endothelial cells and regulation of endothelial barrier function. Histochem. Cell. Biol. 2002, 117, 105–112.
- Vane, J.R.; Anggard, E.E.; Botting, R.M. Regulatory functions of the vascular endothelium. N. Engl. J. Med. 1990, 323, 27–36.
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295.
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837.
- Flavahan, N.A. Atherosclerosis or lipoprotein-induced endothelial dysfunction. Potential mechanisms underlying reduction in EDRF/nitric oxide activity. Circulation 1992, 85, 1927–1938.
- Leeson, P.; Thorne, S.; Donald, A.; Mullen, M.; Clarkson, P.; Deanfield, J. Non-invasive measurement of endothelial function: Effect on brachial artery dilatation of graded endothelial dependent and independent stimuli. Heart 1997, 78, 22–27.
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part I: Basic mechanisms and in vivo monitoring of ROS. Circulation 2003, 108, 1912–1916.
- Benjamin, E.J.; Larson, M.G.; Keyes, M.J.; Mitchell, G.F.; Vasan, R.S.; Keaney, J.F., Jr.; Lehman, B.T.; Fan, S.; Osypiuk, E.; Vita, J.A. Clinical correlates and heritability of flow-mediated dilation in the community: The Framingham Heart Study. Circulation 2004, 109, 613–619.
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27.
- Brook, R.D.; Bard, R.L.; Rubenfire, M.; Ridker, P.M.; Rajagopalan, S. Usefulness of visceral obesity (waist/hip ratio) in predicting vascular endothelial function in healthy overweight adults. Am. J. Cardiol. 2001, 88, 1264–1269.
- Perticone, F.; Ceravolo, R.; Candigliota, M.; Ventura, G.; Iacopino, S.; Sinopoli, F.; Mattioli, P.L. Obesity and body fat distribution induce endothelial dysfunction by oxidative stress: Protective effect of vitamin C. Diabetes 2001, 50, 159–165.
- Arcaro, G.; Zamboni, M.; Rossi, L.; Turcato, E.; Covi, G.; Armellini, F.; Bosello, O.; Lechi, A. Body fat distribution predicts the degree of endothelial dysfunction in uncomplicated obesity. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 936–942.
- McVeigh, G.E.; Brennan, G.M.; Johnston, G.D.; McDermott, B.J.; McGrath, L.T.; Henry, W.R.; Andrews, J.W.; Hayes, J.R. Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 1992, 35, 771–776.
- Williams, S.B.; Cusco, J.A.; Roddy, M.A.; Johnstone, M.T.; Creager, M.A. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Am. Coll. Cardiol. 1996, 27, 567–574.
- Kim, J.A.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal relationships between insulin resistance and endothelial dysfunction: Molecular and pathophysiological mechanisms. Circulation 2006, 113, 1888–1904.
- Fox, C.S. Cardiovascular disease risk factors, type 2 diabetes mellitus, and the Framingham Heart Study. Trends Cardiovasc. Med. 2010, 20, 90–95.
- Avogaro, A.; de Kreutzenberg, S.V.; Fadini, G. Endothelial dysfunction: Causes and consequences in patients with diabetes mellitus. Diabetes Res. Clin. Pract. 2008, 82, S94–S101.
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070.
- Tousoulis, D.; Papageorgiou, N.; Androulakis, E.; Siasos, G.; Latsios, G.; Tentolouris, K.; Stefanadis, C. Diabetes mellitus-associated vascular impairment: Novel circulating biomarkers and therapeutic approaches. J. Am. Coll. Cardiol. 2013, 62, 667–676.
- Arinc, H.; Sarli, B.; Baktir, A.O.; Saglam, H.; Demirci, E.; Dogan, Y.; Kurtul, S.; Karaman, H.; Erden, A.; Karaman, A. Serum gamma glutamyl transferase and alanine transaminase concentrations predict endothelial dysfunction in patients with non-alcoholic steatohepatitis. Ups. J. Med. Sci. 2013, 118, 228–234.
- Villanova, N.; Moscatiello, S.; Ramilli, S.; Bugianesi, E.; Magalotti, D.; Vanni, E.; Zoli, M.; Marchesini, G. Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology 2005, 42, 473–480.
- Colak, Y.; Senates, E.; Yesil, A.; Yilmaz, Y.; Ozturk, O.; Doganay, L.; Coskunpinar, E.; Kahraman, O.T.; Mesci, B.; Ulasoglu, C.; et al. Assessment of endothelial function in patients with nonalcoholic fatty liver disease. Endocrine 2013, 43, 100–107.
- Asahara, T.; Masuda, H.; Takahashi, T.; Kalka, C.; Pastore, C.; Silver, M.; Kearne, M.; Magner, M.; Isner, J.M. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ. Res. 1999, 85, 221–228.
- Murohara, T. Angiogenesis and vasculogenesis for therapeutic neovascularization. Nagoya J. Med. Sci. 2003, 66, 1–7.
- Werner, N.; Junk, S.; Laufs, U.; Link, A.; Walenta, K.; Bohm, M.; Nickenig, G. Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ. Res. 2003, 93, e17–e24.
- Ingram, D.A.; Mead, L.E.; Tanaka, H.; Meade, V.; Fenoglio, A.; Mortell, K.; Pollok, K.; Ferkowicz, M.J.; Gilley, D.; Yoder, M.C. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 2004, 104, 2752–2760.
- Ingram, D.A.; Mead, L.E.; Moore, D.B.; Woodard, W.; Fenoglio, A.; Yoder, M.C. Vessel wall-derived endothelial cells rapidly proliferate because they contain a complete hierarchy of endothelial progenitor cells. Blood 2005, 105, 2783–2786.
- Lin, Y.; Weisdorf, D.J.; Solovey, A.; Hebbel, R.P. Origins of circulating endothelial cells and endothelial outgrowth from blood. J. Clin. Investig. 2000, 105, 71–77.
- Duong, H.T.; Comhair, S.A.; Aldred, M.A.; Mavrakis, L.; Savasky, B.M.; Erzurum, S.C.; Asosingh, K. Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulm. Circ. 2011, 1, 475–486.
- Alphonse, R.S.; Vadivel, A.; Zhong, S.; McConaghy, S.; Ohls, R.; Yoder, M.C.; Thebaud, B. The isolation and culture of endothelial colony-forming cells from human and rat lungs. Nat. Protoc. 2015, 10, 1697–1708.
- Solomon, I.; O’Reilly, M.; Ionescu, L.; Alphonse, R.S.; Rajabali, S.; Zhong, S.; Vadivel, A.; Shelley, W.C.; Yoder, M.C.; Thebaud, B. Functional Differences Between Placental Micro- and Macrovascular Endothelial Colony-Forming Cells. Stem Cells Transl. Med. 2016, 5, 291–300.
- Prasain, N.; Lee, M.R.; Vemula, S.; Meador, J.L.; Yoshimoto, M.; Ferkowicz, M.J.; Fett, A.; Gupta, M.; Rapp, B.M.; Saadatzadeh, M.R.; et al. Differentiation of human pluripotent stem cells to cells similar to cord-blood endothelial colony-forming cells. Nat. Biotechnol. 2014, 32, 1151–1157.
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial Progenitors: A Consensus Statement on Nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320.
- Mund, J.A.; Estes, M.L.; Yoder, M.C.; Ingram, D.A., Jr.; Case, J. Flow cytometric identification and functional characterization of immature and mature circulating endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1045–1053.
- Estes, M.L.; Mund, J.A.; Ingram, D.A.; Case, J. Identification of endothelial cells and progenitor cell subsets in human peripheral blood. Curr. Protoc. Cytom. 2010, 33, 1–11.
- Asahara, T.; Kawamoto, A.; Masuda, H. Concise review: Circulating endothelial progenitor cells for vascular medicine. Stem Cells 2011, 29, 1605–1650.
- Fadini, G.P.; Baesso, I.; Albiero, M.; Sartore, S.; Agostini, C.; Avogaro, A. Technical notes on endothelial progenitor cells: Ways to escape from the knowledge plateau. Atherosclerosis 2008, 197, 496–503.
- Boldicke, T.; Tesar, M.; Griesel, C.; Rohde, M.; Grone, H.J.; Waltenberger, J.; Kollet, O.; Lapidot, T.; Yayon, A.; Weich, H. Anti-VEGFR-2 scFvs for cell isolation. Single-chain antibodies recognizing the human vascular endothelial growth factor receptor-2 (VEGFR-2/flk-1) on the surface of primary endothelial cells and preselected CD34+ cells from cord blood. Stem Cells 2001, 19, 24–36.
- Kunz, G.A.; Liang, G.; Cuculi, F.; Gregg, D.; Vata, K.C.; Shaw, L.K.; Goldschmidt-Clermont, P.J.; Dong, C.; Taylor, D.A.; Peterson, E.D. Circulating endothelial progenitor cells predict coronary artery disease severity. Am. Heart J. 2006, 152, 190–195.
- Keymel, S.; Kalka, C.; Rassaf, T.; Yeghiazarians, Y.; Kelm, M.; Heiss, C. Impaired endothelial progenitor cell function predicts age-dependent carotid intimal thickening. Basic Res. Cardiol. 2008, 103, 582–586.
- Iwasaki, H.; Kawamoto, A.; Ishikawa, M.; Oyamada, A.; Nakamori, S.; Nishimura, H.; Sadamoto, K.; Horii, M.; Matsumoto, T.; Murasawa, S.; et al. Dose-dependent contribution of CD34-positive cell transplantation to concurrent vasculogenesis and cardiomyogenesis for functional regenerative recovery after myocardial infarction. Circulation 2006, 113, 1311–1325.
- Yip, H.K.; Chang, L.T.; Sun, C.K.; Sheu, J.J.; Chiang, C.H.; Youssef, A.A.; Lee, F.Y.; Wu, C.J.; Fu, M. Autologous transplantation of bone marrow-derived endothelial progenitor cells attenuates monocrotaline-induced pulmonary arterial hypertension in rats. Crit. Care Med. 2008, 36, 873–880.
- Yip, H.K.; Chang, L.T.; Chang, W.N.; Lu, C.H.; Liou, C.W.; Lan, M.Y.; Liu, J.S.; Youssef, A.A.; Chang, H.W. Level and value of circulating endothelial progenitor cells in patients after acute ischemic stroke. Stroke 2008, 39, 69–74.
- Berezin, A.E.; Kremzer, A.A.; Berezina, T.A.; Martovitskaya, Y.V. The pattern of circulating microparticles in patients with diabetes mellitus with asymptomatic atherosclerosis. Acta Clin. Belg. 2016, 71, 38–45.
- Bakogiannis, C.; Tousoulis, D.; Androulakis, E.; Briasoulis, A.; Papageorgiou, N.; Vogiatzi, G.; Kampoli, A.M.; Charakida, M.; Siasos, G.; Latsios, G.; et al. Circulating endothelial progenitor cells as biomarkers for prediction of cardiovascular outcomes. Curr. Med. Chem. 2012, 19, 2597–2604.
- Berezin, A.E.; Kremzer, A.A. Circulating endothelial progenitor cells as markers for severity of ischemic chronic heart failure. J. Card. Fail. 2014, 20, 438–447.
- Berezin, A.E.; Kremzer, A.A.; Samura, T.A.; Berezina, T.A.; Martovitskaya, Y.V. Serum uric Acid predicts declining of circulating proangiogenic mononuclear progenitor cells in chronic heart failure patients. J. Cardiovasc. Thorac. Res. 2014, 6, 153–162.
- Egan, C.G.; Lavery, R.; Caporali, F.; Fondelli, C.; Laghi-Pasini, F.; Dotta, F.; Sorrentino, V. Generalised reduction of putative endothelial progenitors and CXCR4-positive peripheral blood cells in type 2 diabetes. Diabetologia 2008, 51, 1296–1305.
- Fadini, G.P. A reappraisal of the role of circulating (progenitor) cells in the pathobiology of diabetic complications. Diabetologia 2014, 57, 4–15.
- Langford-Smith, A.W.W.; Hasan, A.; Weston, R.; Edwards, N.; Jones, A.M.; Boulton, A.J.M.; Bowling, F.L.; Rashid, S.T.; Wilkinson, F.L.; Alexander, M.Y. Diabetic endothelial colony forming cells have the potential for restoration with glycomimetics. Sci. Rep. 2019, 9, 2309.
- Leicht, S.F.; Schwarz, T.M.; Hermann, P.C.; Seissler, J.; Aicher, A.; Heeschen, C. Adiponectin pretreatment counteracts the detrimental effect of a diabetic environment on endothelial progenitors. Diabetes 2011, 60, 652–661.
- Sorrentino, S.A.; Bahlmann, F.H.; Besler, C.; Muller, M.; Schulz, S.; Kirchhoff, N.; Doerries, C.; Horvath, T.; Limbourg, A.; Limbourg, F.; et al. Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus: Restoration by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation 2007, 116, 163–173.
- Tepper, O.M.; Galiano, R.D.; Capla, J.M.; Kalka, C.; Gagne, P.J.; Jacobowitz, G.R.; Levine, J.P.; Gurtner, G.C. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 2002, 106, 2781–2786.
- Ii, M.; Takenaka, H.; Asai, J.; Ibusuki, K.; Mizukami, Y.; Maruyama, K.; Yoon, Y.S.; Wecker, A.; Luedemann, C.; Eaton, E.; et al. Endothelial progenitor thrombospondin-1 mediates diabetes-induced delay in reendothelialization following arterial injury. Circ. Res. 2006, 98, 697–704.
- Oikawa, A.; Siragusa, M.; Quaini, F.; Mangialardi, G.; Katare, R.G.; Caporali, A.; van Buul, J.D.; van Alphen, F.P.; Graiani, G.; Spinetti, G.; et al. Diabetes mellitus induces bone marrow microangiopathy. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 498–508.
- Albiero, M.; Menegazzo, L.; Boscaro, E.; Agostini, C.; Avogaro, A.; Fadini, G.P. Defective recruitment, survival and proliferation of bone marrow-derived progenitor cells at sites of delayed diabetic wound healing in mice. Diabetologia 2011, 54, 945–953.
- Kuliszewski, M.A.; Ward, M.R.; Kowalewski, J.W.; Smith, A.H.; Stewart, D.J.; Kutryk, M.J.; Leong-Poi, H. A direct comparison of endothelial progenitor cell dysfunction in rat metabolic syndrome and diabetes. Atherosclerosis 2013, 226, 58–66.
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625.
- Yue, W.S.; Lau, K.K.; Siu, C.W.; Wang, M.; Yan, G.H.; Yiu, K.H.; Tse, H.F. Impact of glycemic control on circulating endothelial progenitor cells and arterial stiffness in patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2011, 10, 113.
- Fadini, G.P.; Miorin, M.; Facco, M.; Bonamico, S.; Baesso, I.; Grego, F.; Menegolo, M.; de Kreutzenberg, S.V.; Tiengo, A.; Agostini, C.; et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2005, 45, 1449–1457.
- Fadini, G.P.; Sartore, S.; Albiero, M.; Baesso, I.; Murphy, E.; Menegolo, M.; Grego, F.; Vigili de Kreutzenberg, S.; Tiengo, A.; Agostini, C.; et al. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2140–2146.
- Chen, M.C.; Sheu, J.J.; Wang, P.W.; Chen, C.Y.; Kuo, M.C.; Hsieh, C.J.; Chen, J.F.; Chang, H.W. Complications impaired endothelial progenitor cell function in Type 2 diabetic patients with or without critical leg ischaemia: Implication for impaired neovascularization in diabetes. Diabet. Med. 2009, 26, 134–141.
- Moon, J.H.; Chae, M.K.; Kim, K.J.; Kim, H.M.; Cha, B.S.; Lee, H.C.; Kim, Y.J.; Lee, B.W. Decreased endothelial progenitor cells and increased serum glycated albumin are independently correlated with plaque-forming carotid artery atherosclerosis in type 2 diabetes patients without documented ischemic disease. Circ. J. 2012, 76, 2273–2279.
- Chen, Y.H.; Lin, S.J.; Lin, F.Y.; Wu, T.C.; Tsao, C.R.; Huang, P.H.; Liu, P.L.; Chen, Y.L.; Chen, J.W. High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanisms. Diabetes 2007, 56, 1559–1568.
- Werner, N.; Kosiol, S.; Schiegl, T.; Ahlers, P.; Walenta, K.; Link, A.; Bohm, M.; Nickenig, G. Circulating endothelial progenitor cells and cardiovascular outcomes. N. Engl. J. Med. 2005, 353, 999–1007.
- MacEneaney, O.J.; DeSouza, C.A.; Weil, B.R.; Kushner, E.J.; Van Guilder, G.P.; Mestek, M.L.; Greiner, J.J.; Stauffer, B.L. Prehypertension and endothelial progenitor cell function. J. Hum. Hypertens. 2011, 25, 57–62.
- Mandraffino, G.; Imbalzano, E.; Sardo, M.A.; D’Ascola, A.; Mamone, F.; Lo Gullo, A.; Alibrandi, A.; Loddo, S.; Mormina, E.; David, A.; et al. Circulating progenitor cells in hypertensive patients with different degrees of cardiovascular involvement. J. Hum. Hypertens. 2014, 28, 543–550.
- Skrzypkowska, M.W.; Ryba-Stanislawowska, M.E.; Slominski, B.; Gutknecht, P.G.; Siebert, J.; Mysliwska, J.M. Association of circulating progenitor cells with angiotensin II in newly diagnosed hypertensive patients. J. Hum. Hypertens. 2017, 32, 46–53.
- Marketou, M.E.; Kalyva, A.; Parthenakis, F.I.; Pontikoglou, C.; Maragkoudakis, S.; Kontaraki, J.E.; Chlouverakis, G.; Zacharis, E.A.; Patrianakos, A.; Papadaki, H.A.; et al. Circulating endothelial progenitor cells in hypertensive patients with increased arterial stiffness. J. Clin. Hypertens. 2014, 16, 295–300.
- Lewington, S.; Clarke, R.; Qizilbash, N.; Peto, R.; Collins, R.; Prospective Studies, C. Age-specific relevance of usual blood pressure to vascular mortality: A meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002, 360, 1903–1913.
- Britton, K.A.; Gaziano, J.M.; Djousse, L. Normal systolic blood pressure and risk of heart failure in US male physicians. Eur. J. Heart Fail. 2009, 11, 1129–1134.
- Jahangiry, L.; Farhangi, M.A.; Rezaei, F. Framingham risk score for estimation of 10-years of cardiovascular diseases risk in patients with metabolic syndrome. J. Health Popul. Nutr. 2017, 36, 36.
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600.
- Vasa, M.; Fichtlscherer, S.; Aicher, A.; Adler, K.; Urbich, C.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 2001, 89, E1–E7.
- Lee, C.W.; Huang, P.H.; Huang, S.S.; Leu, H.B.; Huang, C.C.; Wu, T.C.; Chen, J.W.; Lin, S.J. Decreased circulating endothelial progenitor cell levels and function in essential hypertensive patients with electrocardiographic left ventricular hypertrophy. Hypertens. Res. 2011, 34, 999–1003.
- Bitterli, L.; Afan, S.; Buhler, S.; DiSanto, S.; Zwahlen, M.; Schmidlin, K.; Yang, Z.; Baumgartner, I.; Diehm, N.; Kalka, C. Endothelial progenitor cells as a biological marker of peripheral artery disease. Vasc. Med. 2016, 21, 3–11.
- Fadini, G.P.; Sartore, S.; Baesso, I.; Lenzi, M.; Agostini, C.; Tiengo, A.; Avogaro, A. Endothelial progenitor cells and the diabetic paradox. Diabetes Care 2006, 29, 714–716.
- Jialal, I.; Devaraj, S.; Singh, U.; Huet, B.A. Decreased number and impaired functionality of endothelial progenitor cells in subjects with metabolic syndrome: Implications for increased cardiovascular risk. Atherosclerosis 2010, 211, 297–302.
- Muller-Ehmsen, J.; Braun, D.; Schneider, T.; Pfister, R.; Worm, N.; Wielckens, K.; Scheid, C.; Frommolt, P.; Flesch, M. Decreased number of circulating progenitor cells in obesity: Beneficial effects of weight reduction. Eur. Heart J. 2008, 29, 1560–1568.
- Heida, N.M.; Muller, J.P.; Cheng, I.F.; Leifheit-Nestler, M.; Faustin, V.; Riggert, J.; Hasenfuss, G.; Konstantinides, S.; Schafer, K. Effects of obesity and weight loss on the functional properties of early outgrowth endothelial progenitor cells. J. Am. Coll. Cardiol. 2010, 55, 357–367.
- Chen, Y.L.; Chang, C.L.; Sun, C.K.; Wu, C.J.; Tsai, T.H.; Chung, S.Y.; Chua, S.; Yeh, K.H.; Leu, S.; Sheu, J.J.; et al. Impact of obesity control on circulating level of endothelial progenitor cells and angiogenesis in response to ischemic stimulation. J. Transl. Med. 2012, 10, 86.
- Tsai, T.H.; Chai, H.T.; Sun, C.K.; Yen, C.H.; Leu, S.; Chen, Y.L.; Chung, S.Y.; Ko, S.F.; Chang, H.W.; Wu, C.J.; et al. Obesity suppresses circulating level and function of endothelial progenitor cells and heart function. J. Transl. Med. 2012, 10, 137.
- Chen, J.Z.; Zhang, F.R.; Tao, Q.M.; Wang, X.X.; Zhu, J.H.; Zhu, J.H. Number and activity of endothelial progenitor cells from peripheral blood in patients with hypercholesterolaemia. Clin. Sci. 2004, 107, 273–280.
- Rossi, F.; Bertone, C.; Montanile, F.; Miglietta, F.; Lubrano, C.; Gandini, L.; Santiemma, V. HDL cholesterol is a strong determinant of endothelial progenitor cells in hypercholesterolemic subjects. Microvasc. Res. 2010, 80, 274–279.
- Imanishi, T.; Hano, T.; Sawamura, T.; Nishio, I. Oxidized low-density lipoprotein induces endothelial progenitor cell senescence, leading to cellular dysfunction. Clin. Exp. Pharmacol. Physiol. 2004, 31, 407–413.
- Tarantino, G.; Saldalamacchia, G.; Conca, P.; Arena, A. Non-alcoholic fatty liver disease: Further expression of the metabolic syndrome. J. Gastroenterol. Hepatol. 2007, 22, 293–303.
- Kotronen, A.; Yki-Jarvinen, H. Fatty liver: A novel component of the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 27–38.
- Despres, J.P.; Lemieux, I.; Bergeron, J.; Pibarot, P.; Mathieu, P.; Larose, E.; Rodes-Cabau, J.; Bertrand, O.F.; Poirier, P. Abdominal obesity and the metabolic syndrome: Contribution to global cardiometabolic risk. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1039–1049.
- Vlachopoulos, C.; Manesis, E.; Baou, K.; Papatheodoridis, G.; Koskinas, J.; Tiniakos, D.; Aznaouridis, K.; Archimandritis, A.; Stefanadis, C. Increased arterial stiffness and impaired endothelial function in nonalcoholic Fatty liver disease: A pilot study. Am. J. Hypertens. 2010, 23, 1183–1189.
- Chiang, C.H.; Huang, P.H.; Chung, F.P.; Chen, Z.Y.; Leu, H.B.; Huang, C.C.; Wu, T.C.; Chen, J.W.; Lin, S.J. Decreased circulating endothelial progenitor cell levels and function in patients with nonalcoholic fatty liver disease. PLoS ONE 2012, 7, e31799.
- Gutierrez-Grobe, Y.; Gavilanes-Espinar, J.G.; Masso-Rojas, F.A.; Sanchez-Valle, V.; Paez-Arenas, A.; Ponciano-Rodriguez, G.; Chavez-Tapia, N.C.; Uribe, M.; Mendez-Sanchez, N. Metabolic syndrome and nonalcoholic fatty liver disease. The role of endothelial progenitor cells. Ann. Hepatol. 2013, 12, 908–914.
- Dean, R.T.; Fu, S.; Stocker, R.; Davies, M.J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 1997, 324, 1–18.
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209.
- Versari, D.; Daghini, E.; Virdis, A.; Ghiadoni, L.; Taddei, S. Endothelium-dependent contractions and endothelial dysfunction in human hypertension. Br. J. Pharmacol. 2009, 157, 527–536.
- Watson, T.; Goon, P.K.; Lip, G.Y. Endothelial progenitor cells, endothelial dysfunction, inflammation, and oxidative stress in hypertension. Antioxid. Redox Signal. 2008, 10, 1079–1088.
- Schatteman, G.C.; Hanlon, H.D.; Jiao, C.; Dodds, S.G.; Christy, B.A. Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J. Clin. Investig. 2000, 106, 571–578.
- Thum, T.; Fraccarollo, D.; Galuppo, P.; Tsikas, D.; Frantz, S.; Ertl, G.; Bauersachs, J. Bone marrow molecular alterations after myocardial infarction: Impact on endothelial progenitor cells. Cardiovasc. Res. 2006, 70, 50–60.
- Dernbach, E.; Urbich, C.; Brandes, R.P.; Hofmann, W.K.; Zeiher, A.M.; Dimmeler, S. Antioxidative stress-associated genes in circulating progenitor cells: Evidence for enhanced resistance against oxidative stress. Blood 2004, 104, 3591–3597.
- Collett, J.A.; Mehrotra, P.; Crone, A.; Shelley, W.C.; Yoder, M.C.; Basile, D.P. Endothelial colony-forming cells ameliorate endothelial dysfunction via secreted factors following ischemia-reperfusion injury. Am. J. Physiol. Renal. Physiol. 2017, 312, F897–F907.
- Tao, J.; Yang, Z.; Wang, J.M.; Wang, L.C.; Luo, C.F.; Tang, A.L.; Dong, Y.G.; Ma, H. Shear stress increases Cu/Zn SOD activity and mRNA expression in human endothelial progenitor cells. J. Hum. Hypertens. 2007, 21, 353–358.
- He, T.; Peterson, T.E.; Holmuhamedov, E.L.; Terzic, A.; Caplice, N.M.; Oberley, L.W.; Katusic, Z.S. Human endothelial progenitor cells tolerate oxidative stress due to intrinsically high expression of manganese superoxide dismutase. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2021–2027.
- Lin, F.Y.; Tsao, N.W.; Shih, C.M.; Lin, Y.W.; Yeh, J.S.; Chen, J.W.; Nakagami, H.; Morishita, R.; Sawamura, T.; Huang, C.Y. The biphasic effects of oxidized-low density lipoprotein on the vasculogenic function of endothelial progenitor cells. PLoS ONE 2015, 10, e0123971.
- Tso, C.; Martinic, G.; Fan, W.H.; Rogers, C.; Rye, K.A.; Barter, P.J. High-density lipoproteins enhance progenitor-mediated endothelium repair in mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1144–1149.
- Beckman, K.B.; Ames, B.N. Endogenous oxidative damage of mtDNA. Mutat. Res. 1999, 424, 51–58.
- Murphy, M.P. Induction of mitochondrial ROS production by electrophilic lipids: A new pathway of redox signaling? Am. J. Physiol. Heart. Circ. Physiol. 2006, 290, H1754–H1755.
- Choi, J.W.; Son, S.M.; Mook-Jung, I.; Moon, Y.J.; Lee, J.Y.; Wang, K.C.; Kang, H.S.; Phi, J.H.; Choi, S.A.; Chong, S.; et al. Mitochondrial abnormalities related to the dysfunction of circulating endothelial colony-forming cells in moyamoya disease. J. Neurosurg. 2018, 129, 1151–1159.
- Lyons, C.J.; O’Brien, T. The Functionality of Endothelial-Colony-Forming Cells from Patients with Diabetes Mellitus. Cells 2020, 9, 1731.
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2013, 5, 37–50.
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell. Biol. 2007, 8, 729–740.
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705.
- Martin-Ruiz, C.; Saretzki, G.; Petrie, J.; Ladhoff, J.; Jeyapalan, J.; Wei, W.; Sedivy, J.; von Zglinicki, T. Stochastic variation in telomere shortening rate causes heterogeneity of human fibroblast replicative life span. J. Biol. Chem. 2004, 279, 17826–17833.
- Vassallo, P.F.; Simoncini, S.; Ligi, I.; Chateau, A.L.; Bachelier, R.; Robert, S.; Morere, J.; Fernandez, S.; Guillet, B.; Marcelli, M.; et al. Accelerated senescence of cord blood endothelial progenitor cells in premature neonates is driven by SIRT1 decreased expression. Blood 2014, 123, 2116–2126.
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118.
- Ingram, D.A.; Lien, I.Z.; Mead, L.E.; Estes, M.; Prater, D.N.; Derr-Yellin, E.; DiMeglio, L.A.; Haneline, L.S. In vitro hyperglycemia or a diabetic intrauterine environment reduces neonatal endothelial colony-forming cell numbers and function. Diabetes 2008, 57, 724–731.
- Carracedo, J.; Merino, A.; Briceno, C.; Soriano, S.; Buendia, P.; Calleros, L.; Rodriguez, M.; Martin-Malo, A.; Aljama, P.; Ramirez, R. Carbamylated low-density lipoprotein induces oxidative stress and accelerated senescence in human endothelial progenitor cells. FASEB J. 2011, 25, 1314–1322.
- Satoh, M.; Ishikawa, Y.; Takahashi, Y.; Itoh, T.; Minami, Y.; Nakamura, M. Association between oxidative DNA damage and telomere shortening in circulating endothelial progenitor cells obtained from metabolic syndrome patients with coronary artery disease. Atherosclerosis 2008, 198, 347–353.
- Alphonse, R.S.; Vadivel, A.; Fung, M.; Shelley, W.C.; Critser, P.J.; Ionescu, L.; O’Reilly, M.; Ohls, R.K.; McConaghy, S.; Eaton, F.; et al. Existence, functional impairment, and lung repair potential of endothelial colony-forming cells in oxygen-induced arrested alveolar growth. Circulation 2014, 129, 2144–2157.
- Lee, S.H.; Lee, J.H.; Han, Y.S.; Ryu, J.M.; Yoon, Y.M.; Han, H.J. Hypoxia accelerates vascular repair of endothelial colony-forming cells on ischemic injury via STAT3-BCL3 axis. Stem Cell Res. Ther. 2015, 6, 139.
- Tsukada, S.; Kwon, S.M.; Matsuda, T.; Jung, S.Y.; Lee, J.H.; Lee, S.H.; Masuda, H.; Asahara, T. Identification of mouse colony-forming endothelial progenitor cells for postnatal neovascularization: A novel insight highlighted by new mouse colony-forming assay. Stem Cell Res. Ther. 2013, 4, 20.
- Luque Contreras, D.; Vargas Robles, H.; Romo, E.; Rios, A.; Escalante, B. The role of nitric oxide in the post-ischemic revascularization process. Pharmacol. Ther. 2006, 112, 553–563.
- Duda, D.G.; Fukumura, D.; Jain, R.K. Role of eNOS in neovascularization: NO for endothelial progenitor cells. Trends Mol. Med. 2004, 10, 143–145.
- Aicher, A.; Heeschen, C.; Mildner-Rihm, C.; Urbich, C.; Ihling, C.; Technau-Ihling, K.; Zeiher, A.M.; Dimmeler, S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat. Med. 2003, 9, 1370–1376.
- Babaei, S.; Stewart, D.J. Overexpression of endothelial NO synthase induces angiogenesis in a co-culture model. Cardiovasc. Res. 2002, 55, 190–200.
- Aicher, A.; Zeiher, A.M.; Dimmeler, S. Mobilizing endothelial progenitor cells. Hypertension 2005, 45, 321–325.
- Heissig, B.; Werb, Z.; Rafii, S.; Hattori, K. Role of c-kit/Kit ligand signaling in regulating vasculogenesis. Thromb. Haemost. 2003, 90, 570–576.
- Thum, T.; Fraccarollo, D.; Schultheiss, M.; Froese, S.; Galuppo, P.; Widder, J.D.; Tsikas, D.; Ertl, G.; Bauersachs, J. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes 2007, 56, 666–674.
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605.
- Vasa, M.; Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ. Res. 2000, 87, 540–542.
- Hoetzer, G.L.; Irmiger, H.M.; Keith, R.S.; Westbrook, K.M.; DeSouza, C.A. Endothelial nitric oxide synthase inhibition does not alter endothelial progenitor cell colony forming capacity or migratory activity. J. Cardiovasc. Pharmacol. 2005, 46, 387–389.
- Li Calzi, S.; Purich, D.L.; Chang, K.H.; Afzal, A.; Nakagawa, T.; Busik, J.V.; Agarwal, A.; Segal, M.S.; Grant, M.B. Carbon monoxide and nitric oxide mediate cytoskeletal reorganization in microvascular cells via vasodilator-stimulated phosphoprotein phosphorylation: Evidence for blunted responsiveness in diabetes. Diabetes 2008, 57, 2488–2494.
- Segal, M.S.; Shah, R.; Afzal, A.; Perrault, C.M.; Chang, K.; Schuler, A.; Beem, E.; Shaw, L.C.; Li Calzi, S.; Harrison, J.K.; et al. Nitric oxide cytoskeletal-induced alterations reverse the endothelial progenitor cell migratory defect associated with diabetes. Diabetes 2006, 55, 102–109.
- Chen, Y.S.; Chen, K.H.; Liu, C.C.; Lee, C.T.; Yang, C.H.; Chuang, K.C.; Lin, C.R. Propofol-induced vascular permeability change is related to the nitric oxide signaling pathway and occludin phosphorylation. J. Biomed. Sci. 2007, 14, 629–636.
- Verma, S.; Szmitko, P.E.; Anderson, T.J. Endothelial function: Ready for prime time? Can. J. Cardiol. 2004, 20, 1335–1339.
- Kalka, C.; Masuda, H.; Takahashi, T.; Gordon, R.; Tepper, O.; Gravereaux, E.; Pieczek, A.; Iwaguro, H.; Hayashi, S.I.; Isner, J.M.; et al. Vascular endothelial growth factor(165) gene transfer augments circulating endothelial progenitor cells in human subjects. Circ. Res. 2000, 86, 1198–1202.
- Young, P.P.; Hofling, A.A.; Sands, M.S. VEGF increases engraftment of bone marrow-derived endothelial progenitor cells (EPCs) into vasculature of newborn murine recipients. Proc. Natl. Acad. Sci. USA 2002, 99, 11951–11956.
- Dulak, J.; Jozkowicz, A.; Frick, M.; Alber, H.F.; Dichtl, W.; Schwarzacher, S.P.; Pachinger, O.; Weidinger, F. Vascular endothelial growth factor: Angiogenesis, atherogenesis or both? J. Am. Coll. Cardiol. 2001, 38, 2137–2138.
- Dulak, J.; Jozkowicz, A.; Dembinska-Kiec, A.; Guevara, I.; Zdzienicka, A.; Zmudzinska-Grochot, D.; Florek, I.; Wojtowicz, A.; Szuba, A.; Cooke, J.P. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 659–666.
- Kimura, H.; Esumi, H. Reciprocal regulation between nitric oxide and vascular endothelial growth factor in angiogenesis. Acta Biochim. Pol. 2003, 50, 49–59.
- Di, Y.; Zhang, D.; Hu, T.; Li, D. miR-23 regulate the pathogenesis of patients with coronary artery disease. Int J. Clin. Exp. Med. 2015, 8, 11759–11769.
- Qin, W.; Xie, W.; Xia, N.; He, Q.; Sun, T. Silencing of Transient Receptor Potential Channel 4 Alleviates oxLDL-induced Angiogenesis in Human Coronary Artery Endothelial Cells by Inhibition of VEGF and NF-kappaB. Med. Sci. Monit. 2016, 22, 930–936.
- Li, H.; Zhang, X.; Guan, X.; Cui, X.; Wang, Y.; Chu, H.; Cheng, M. Advanced glycation end products impair the migration, adhesion and secretion potentials of late endothelial progenitor cells. Cardiovasc. Diabetol. 2012, 11, 46.
- Edwards, N.; Langford-Smith, A.W.W.; Wilkinson, F.L.; Alexander, M.Y. Endothelial Progenitor Cells: New Targets for Therapeutics for Inflammatory Conditions With High Cardiovascular Risk. Front. Med. 2018, 5, 200.
- Castejon, R.; Jimenez-Ortiz, C.; Rosado, S.; Tutor-Ureta, P.; Mellor-Pita, S.; Yebra-Bango, M. Metabolic syndrome is associated with decreased circulating endothelial progenitor cells and increased arterial stiffness in systemic lupus erythematosus. Lupus 2016, 25, 129–136.
- Mok, C.C. Metabolic syndrome and systemic lupus erythematosus: The connection. Expert Rev. Clin. Immunol. 2019, 15, 765–775.
- Rodriguez-Carrio, J.; Prado, C.; de Paz, B.; Lopez, P.; Gomez, J.; Alperi-Lopez, M.; Ballina-Garcia, F.J.; Suarez, A. Circulating endothelial cells and their progenitors in systemic lupus erythematosus and early rheumatoid arthritis patients. Rheumatology 2012, 51, 1775–1784.
- Rodriguez-Carrio, J.; de Paz, B.; Lopez, P.; Prado, C.; Alperi-Lopez, M.; Ballina-Garcia, F.J.; Suarez, A. IFNalpha serum levels are associated with endothelial progenitor cells imbalance and disease features in rheumatoid arthritis patients. PLoS ONE 2014, 9, e86069.
- Lee, P.Y.; Li, Y.; Richards, H.B.; Chan, F.S.; Zhuang, H.; Narain, S.; Butfiloski, E.J.; Sobel, E.S.; Reeves, W.H.; Segal, M.S. Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 3759–3769.
- Thacker, S.G.; Zhao, W.; Smith, C.K.; Luo, W.; Wang, H.; Vivekanandan-Giri, A.; Rabquer, B.J.; Koch, A.E.; Pennathur, S.; Davidson, A.; et al. Type I interferons modulate vascular function, repair, thrombosis, and plaque progression in murine models of lupus and atherosclerosis. Arthritis Rheum. 2012, 64, 2975–2985.
- Kahlenberg, J.M.; Thacker, S.G.; Berthier, C.C.; Cohen, C.D.; Kretzler, M.; Kaplan, M.J. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J. Immunol. 2011, 187, 6143–6156.
- Chen, T.G.; Zhong, Z.Y.; Sun, G.F.; Zhou, Y.X.; Zhao, Y. Effects of tumour necrosis factor-alpha on activity and nitric oxide synthase of endothelial progenitor cells from peripheral blood. Cell Prolif. 2011, 44, 352–359.
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864.
- Fadini, G.P.; Madeddu, P.; Waltenberger, J.; Fiorina, P. Vascular stem and progenitor cells in diabetic complications. Exp. Diabetes Res. 2012, 2012, 580343.
- Fadini, G.P.; Sartore, S.; Schiavon, M.; Albiero, M.; Baesso, I.; Cabrelle, A.; Agostini, C.; Avogaro, A. Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia-reperfusion injury in rats. Diabetologia 2006, 49, 3075–3084.
- Fadini, G.P.; Avogaro, A. Diabetes impairs mobilization of stem cells for the treatment of cardiovascular disease: A meta-regression analysis. Int. J. Cardiol. 2013, 168, 892–897.
- Gallagher, K.A.; Liu, Z.J.; Xiao, M.; Chen, H.; Goldstein, L.J.; Buerk, D.G.; Nedeau, A.; Thom, S.R.; Velazquez, O.C. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J. Clin. Investig. 2007, 117, 1249–1259.
- Barwari, T.; Rienks, M.; Mayr, M. MicroRNA-21 and the Vulnerability of Atherosclerotic Plaques. Mol. Ther. 2018, 26, 938–940.
- Navickas, R.; Gal, D.; Laucevicius, A.; Taparauskaite, A.; Zdanyte, M.; Holvoet, P. Identifying circulating microRNAs as biomarkers of cardiovascular disease: A systematic review. Cardiovasc. Res. 2016, 111, 322–337.
- Qu, K.; Wang, Z.; Lin, X.L.; Zhang, K.; He, X.L.; Zhang, H. MicroRNAs: Key regulators of endothelial progenitor cell functions. Clin. Chim. Acta 2015, 448, 65–73.
- Ito, T.; Yagi, S.; Yamakuchi, M. MicroRNA-34a regulation of endothelial senescence. Biochem. Biophys. Res. Commun. 2010, 398, 735–740.
- Santulli, G. MicroRNAs and Endothelial (Dys) Function. J. Cell. Physiol. 2016, 231, 1638–1644.
- Meng, S.; Cao, J.T.; Zhang, B.; Zhou, Q.; Shen, C.X.; Wang, C.Q. Downregulation of microRNA-126 in endothelial progenitor cells from diabetes patients, impairs their functional properties, via target gene Spred-1. J. Mol. Cell. Cardiol. 2012, 53, 64–72.
- Meng, S.; Cao, J.; Zhang, X.; Fan, Y.; Fang, L.; Wang, C.; Lv, Z.; Fu, D.; Li, Y. Downregulation of microRNA-130a contributes to endothelial progenitor cell dysfunction in diabetic patients via its target Runx3. PLoS ONE 2013, 8, e68611.
- Wang, Y.; Zuo, Q.; Bi, Y.; Zhang, W.; Jin, J.; Zhang, L.; Zhang, Y.N.; Li, B. miR-31 Regulates Spermatogonial Stem Cells Meiosis via Targeting Stra8. J. Cell. Biochem. 2017, 118, 4844–4853.
- Lu, W.C.; Liu, C.J.; Tu, H.F.; Chung, Y.T.; Yang, C.C.; Kao, S.Y.; Chang, K.W.; Lin, S.C. miR-31 targets ARID1A and enhances the oncogenicity and stemness of head and neck squamous cell carcinoma. Oncotarget 2016, 7, 57254–57267.
- Wang, H.W.; Huang, T.S.; Lo, H.H.; Huang, P.H.; Lin, C.C.; Chang, S.J.; Liao, K.H.; Tsai, C.H.; Chan, C.H.; Tsai, C.F.; et al. Deficiency of the microRNA-31-microRNA-720 pathway in the plasma and endothelial progenitor cells from patients with coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 857–869.
- Zhao, T.; Li, J.; Chen, A.F. MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E110–E116.
- Lian, W.; Hu, X.; Shi, R.; Han, S.; Cao, C.; Wang, K.; Li, M. MiR-31 regulates the function of diabetic endothelial progenitor cells by targeting Satb2. Acta Biochim. Biophys. Sin. 2018, 50, 336–344.
- Choi, S.W.; Friso, S. Epigenetics: A New Bridge between Nutrition and Health. Adv. Nutr. 2010, 1, 8–16.
- Wang, C.; Wang, F.; Li, Z.; Cao, Q.; Huang, L.; Chen, S. MeCP2-mediated epigenetic regulation in senescent endothelial progenitor cells. Stem Cell Res. Ther. 2018, 9, 87.
- Fraineau, S.; Palii, C.G.; McNeill, B.; Ritso, M.; Shelley, W.C.; Prasain, N.; Chu, A.; Vion, E.; Rieck, K.; Nilufar, S.; et al. Epigenetic Activation of Pro-angiogenic Signaling Pathways in Human Endothelial Progenitors Increases Vasculogenesis. Stem Cell Rep. 2017, 9, 1573–1587.
- Ohtani, K.; Vlachojannis, G.J.; Koyanagi, M.; Boeckel, J.N.; Urbich, C.; Farcas, R.; Bonig, H.; Marquez, V.E.; Zeiher, A.M.; Dimmeler, S. Epigenetic regulation of endothelial lineage committed genes in pro-angiogenic hematopoietic and endothelial progenitor cells. Circ. Res. 2011, 109, 1219–1229.
- McCully, K.S. Homocysteine and vascular disease. Nat. Med. 1996, 2, 386–389.
- Kang, S.S.; Wong, P.W.; Malinow, M.R. Hyperhomocyst(e)inemia as a risk factor for occlusive vascular disease. Annu. Rev. Nutr. 1992, 12, 279–298.
- Welch, G.N.; Loscalzo, J. Homocysteine and atherothrombosis. N. Engl. J. Med. 1998, 338, 1042–1050.
- Rasmussen, K.; Moller, J. Total homocysteine measurement in clinical practice. Ann. Clin. Biochem. 2000, 37, 627–648.
- Yang, Q.; He, G.W. Imbalance of Homocysteine and H2S: Significance, Mechanisms, and Therapeutic Promise in Vascular Injury. Oxid. Med. Cell. Longev. 2019, 2019, 7629673.
- Citi, V.; Martelli, A.; Gorica, E.; Brogi, S.; Testai, L.; Calderone, V. Role of hydrogen sulfide in endothelial dysfunction: Pathophysiology and therapeutic approaches. J. Adv. Res. 2021, 27, 99–113.
- Stampfer, M.J.; Malinow, M.R. Can lowering homocysteine levels reduce cardiovascular risk? N. Engl. J. Med. 1995, 332, 328–329.
- Dawson, H.; Collins, G.; Pyle, R.; Deep-Dixit, V.; Taub, D.D. The immunoregulatory effects of homocysteine and its intermediates on T-lymphocyte function. Mech. Ageing Dev. 2004, 125, 107–110.
- Selhub, J.; Jacques, P.F.; Wilson, P.W.; Rush, D.; Rosenberg, I.H. Vitamin status and intake as primary determinants of homocysteinemia in an elderly population. JAMA 1993, 270, 2693–2698.
- Refsum, H.; Smith, A.D.; Ueland, P.M.; Nexo, E.; Clarke, R.; McPartlin, J.; Johnston, C.; Engbaek, F.; Schneede, J.; McPartlin, C.; et al. Facts and recommendations about total homocysteine determinations: An expert opinion. Clin. Chem. 2004, 50, 3–32.
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6.
- Lentz, S.R.; Erger, R.A.; Dayal, S.; Maeda, N.; Malinow, M.R.; Heistad, D.D.; Faraci, F.M. Folate dependence of hyperhomocysteinemia and vascular dysfunction in cystathionine beta-synthase-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H970–H975.
- Ungvari, Z.; Pacher, P.; Rischak, K.; Szollar, L.; Koller, A. Dysfunction of nitric oxide mediation in isolated rat arterioles with methionine diet-induced hyperhomocysteinemia. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1899–1904.
- Lentz, S.R.; Sobey, C.G.; Piegors, D.J.; Bhopatkar, M.Y.; Faraci, F.M.; Malinow, M.R.; Heistad, D.D. Vascular dysfunction in monkeys with diet-induced hyperhomocyst(e)inemia. J. Clin. Investig. 1996, 98, 24–29.
- Widner, B.; Enzinger, C.; Laich, A.; Wirleitner, B.; Fuchs, D. Hyperhomocysteinemia, pteridines and oxidative stress. Curr. Drug Metab. 2002, 3, 225–232.
- Alvarez, B.; Demicheli, V.; Duran, R.; Trujillo, M.; Cervenansky, C.; Freeman, B.A.; Radi, R. Inactivation of human Cu, Zn superoxide dismutase by peroxynitrite and formation of histidinyl radical. Free Radic. Biol. Med. 2004, 37, 813–822.
- MacMillan-Crow, L.A.; Crow, J.P.; Thompson, J.A. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 1998, 37, 1613–1622.
- Tyagi, N.; Sedoris, K.C.; Steed, M.; Ovechkin, A.V.; Moshal, K.S.; Tyagi, S.C. Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2649–H2656.
- Mayo, J.N.; Beard, R.S., Jr.; Price, T.O.; Chen, C.H.; Erickson, M.A.; Ercal, N.; Banks, W.A.; Bearden, S.E. Nitrative stress in cerebral endothelium is mediated by mGluR5 in hyperhomocysteinemia. J. Cereb. Blood Flow Metab. 2012, 32, 825–834.
- Calcerrada, P.; Peluffo, G.; Radi, R. Nitric oxide-derived oxidants with a focus on peroxynitrite: Molecular targets, cellular responses and therapeutic implications. Curr. Pharm. Des. 2011, 17, 3905–3932.
- Szabo, C.; Mabley, J.G.; Moeller, S.M.; Shimanovich, R.; Pacher, P.; Virag, L.; Soriano, F.G.; Van Duzer, J.H.; Williams, W.; Salzman, A.L.; et al. Part I: Pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: Studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol. Med. 2002, 8, 571–580.
- Chen, J.Z.; Zhu, J.H.; Wang, X.X.; Zhu, J.H.; Xie, X.D.; Sun, J.; Shang, Y.P.; Guo, X.G.; Dai, H.M.; Hu, S.J. Effects of homocysteine on number and activity of endothelial progenitor cells from peripheral blood. J. Mol. Cell. Cardiol. 2004, 36, 233–239.
- Zhu, J.; Wang, X.; Chen, J.; Sun, J.; Zhang, F. Reduced number and activity of circulating endothelial progenitor cells from patients with hyperhomocysteinemia. Arch. Med. Res. 2006, 37, 484–489.
- Dong, Y.; Sun, Q.; Liu, T.; Wang, H.; Jiao, K.; Xu, J.; Liu, X.; Liu, H.; Wang, W. Nitrative Stress Participates in Endothelial Progenitor Cell Injury in Hyperhomocysteinemia. PLoS ONE 2016, 11, e0158672.
- Alam, M.M.; Mohammad, A.A.; Shuaib, U.; Wang, C.; Ghani, U.; Schwindt, B.; Todd, K.G.; Shuaib, A. Homocysteine reduces endothelial progenitor cells in stroke patients through apoptosis. J. Cereb. Blood Flow Metab. 2009, 29, 157–165.
- Noor, R.; Shuaib, U.; Wang, C.X.; Todd, K.; Ghani, U.; Schwindt, B.; Shuaib, A. High-density lipoprotein cholesterol regulates endothelial progenitor cells by increasing eNOS and preventing apoptosis. Atherosclerosis 2007, 192, 92–99.
- Behera, J.; Tyagi, S.C.; Tyagi, N. Hyperhomocysteinemia induced endothelial progenitor cells dysfunction through hyper-methylation of CBS promoter. Biochem. Biophys. Res. Commun. 2019, 510, 135–141.
- Polhemus, D.J.; Lefer, D.J. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 2014, 114, 730–737.
- Ciccone, V.; Genah, S.; Morbidelli, L. Endothelium as a Source and Target of H2S to Improve Its Trophism and Function. Antioxidants 2021, 10, 486.
- Paul, B.D.; Snyder, S.H. H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700.
- Beltowski, J. Synthesis, Metabolism, and Signaling Mechanisms of Hydrogen Sulfide: An Overview. Methods Mol. Biol. 2019, 2007, 1–8.
- Cheng, Z.; Garikipati, V.N.; Nickoloff, E.; Wang, C.; Polhemus, D.J.; Zhou, J.; Benedict, C.; Khan, M.; Verma, S.K.; Rabinowitz, J.E.; et al. Restoration of Hydrogen Sulfide Production in Diabetic Mice Improves Reparative Function of Bone Marrow Cells. Circulation 2016, 134, 1467–1483.
- Cai, W.J.; Wang, M.J.; Moore, P.K.; Jin, H.M.; Yao, T.; Zhu, Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 2007, 76, 29–40.
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977.
- Szabo, C.; Papapetropoulos, A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br. J. Pharmacol. 2011, 164, 853–865.
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001, 20, 6008–6016.
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol. Cell. 2012, 45, 13–24.
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919.
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Modis, K.; Panopoulos, P.; Asimakopoulou, A.; Gero, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166.
- Altaany, Z.; Ju, Y.; Yang, G.; Wang, R. The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci. Signal. 2014, 7, ra87.
- Altaany, Z.; Yang, G.; Wang, R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J. Cell. Mol. Med. 2013, 17, 879–888.
- Liu, F.; Chen, D.D.; Sun, X.; Xie, H.H.; Yuan, H.; Jia, W.; Chen, A.F. Hydrogen sulfide improves wound healing via restoration of endothelial progenitor cell functions and activation of angiopoietin-1 in type 2 diabetes. Diabetes 2014, 63, 1763–1778.
- Obi, S.; Yamamoto, K.; Ando, J. Effects of shear stress on endothelial progenitor cells. J. Biomed. Nanotechnol. 2014, 10, 2586–2597.
- Obi, S.; Masuda, H.; Shizuno, T.; Sato, A.; Yamamoto, K.; Ando, J.; Abe, Y.; Asahara, T. Fluid shear stress induces differentiation of circulating phenotype endothelial progenitor cells. Am. J. Physiol. Cell. Physiol. 2012, 303, C595–C606.
- Xia, W.H.; Yang, Z.; Xu, S.Y.; Chen, L.; Zhang, X.Y.; Li, J.; Liu, X.; Qiu, Y.X.; Shuai, X.T.; Tao, J. Age-related decline in reendothelialization capacity of human endothelial progenitor cells is restored by shear stress. Hypertension 2012, 59, 1225–1231.
- Huang, B.; Chen, C.T.; Chen, C.S.; Wang, Y.M.; Hsieh, H.J.; Wang, D.L. Laminar shear flow increases hydrogen sulfide and activates a nitric oxide producing signaling cascade in endothelial cells. Biochem. Biophys. Res. Commun. 2015, 464, 1254–1259.
- Hu, Q.; Zhang, B.; Liu, Y.; Guo, Y.; Zhang, T.; Nie, R.; Ke, X.; Dong, X. The effect of fluid shear stress in hydrogen sulphide production and cystathionine gamma-lyase expression in human early endothelial progenitor cells. Ann. Transl. Med. 2020, 8, 1318.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
06 Jul 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No