 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Youjun Zhang | + 2486 word(s) | 2486 | 2021-07-05 05:20:31 | | | |
| 2 | Peter Tang | Meta information modification | 2486 | 2021-07-06 03:32:27 | | |
Video Upload Options
The study of protein–protein interactions (PPIs) is fundamental in understanding the unique role of proteins within cells and their contribution to complex biological systems. Affinity purification coupled to mass spectrometry (AP-MS) and proximity labeling coupled to mass spectrometry (PL-MS) are two powerful techniques that have significantly enhanced our understanding of PPIs. Relying on the specific binding properties of a protein to an immobilized ligand, AP is a fast, sensitive and targeted approach used to detect interactions between bait (protein of interest) and prey (interacting partners) under near-physiological conditions.
1. Introduction
2. Affinity Purification Mass Spectrometry in Plants
|
Tag |
Sequence/Size |
Affinity Resin |
Elution Conditions |
Reference |
|---|---|---|---|---|
|
TAPi tag |
45 kDa |
Calmodulin binding peptide with two protein A domain |
Protein A/low pH |
|
|
Streptavidin binding peptide (SBP) |
WSHPQFEK |
Streptavidin |
Desthiobiotin |
|
|
GSyellow |
37 kDa |
Streptavidin-binding peptide tag with citrine yellow fluorescent protein |
Desthiobiotin/pH |
|
|
Fluorescent protein (GFP, YFP) |
26.9 kDa |
Anti-GFP |
pH |
|
|
GSrhino tag |
21.9 kDa |
two IgG-binding domains of protein G and a SBP tag |
Streptavidin elution buffer [5] |
|
|
Alternative TAP (TAPa) |
26 kDa |
2 xIgG-BD with 6 XHis and 9 Xmyc |
HR3C cleavage/Imidazole/low pH |
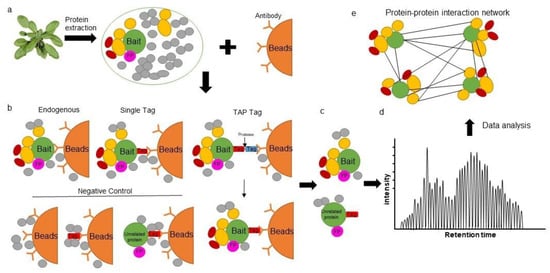
3. The Proximity Labeling Method
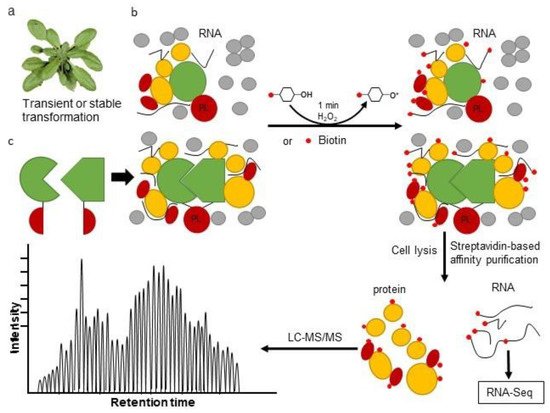
4. Combining Proximity Labeling and Affinity Purification-Mass Spectrometry
References
- Zhang, Y.; Fernie, A.R. Stable and temporary enzyme complexes and metabolons involved in energy and redox metabolism. Antioxid. Redox Signal. 2020.
- Chae, L.; Lee, I.; Shin, J.; Rhee, S.Y. Towards understanding how molecular networks evolve in plants. Curr. Opin. Plant Biol. 2012, 15, 177–184.
- Lampugnani, E.R.; Wink, R.H.; Persson, S.; Somssich, M. The toolbox to study protein–protein interactions in plants. Crit. Rev. Plant Sci. 2018, 37, 308–334.
- Struk, S.; Jacobs, A.; Sánchez Martín-Fontecha, E.; Gevaert, K.; Cubas, P.; Goormachtig, S. Exploring the protein–protein interaction landscape in plants. Plant Cell Environ. 2019, 42, 387–409.
- Van Leene, J.; Eeckhout, D.; Cannoot, B.; De Winne, N.; Persiau, G.; Van De Slijke, E.; Vercruysse, L.; Dedecker, M.; Verkest, A.; Vandepoele, K.; et al. An improved toolbox to unravel the plant cellular machinery by tandem affinity purification of arabidopsis protein complexes. Nat. Protoc. 2015, 10, 169–187.
- Dunham, W.H.; Mullin, M.; Gingras, A.C. Affinity-purification coupled to mass spectrometry: Basic principles and strategies. Proteomics 2012, 12, 1576–1590.
- Zhang, Y.; Gao, P.; Yuan, J.S. Plant protein-protein interaction network and interactome. Curr. Genom. 2010, 11, 40–46.
- Dong, S.; Lau, V.; Song, R.; Ierullo, M.; Esteban, E.; Wu, Y.; Sivieng, T.; Nahal, H.; Gaudinier, A.; Pasha, A. Proteome-wide, structure-based prediction of protein-protein interactions/new molecular interactions viewer. Plant Physiol. 2019, 179, 1893–1907.
- Jiang, M.; Niu, C.; Cao, J.; Ni, D.A.; Chu, Z. In silico-prediction of protein–protein interactions network about mapks and pp2cs reveals a novel docking site variants in brachypodium distachyon. Sci. Rep. 2018, 8, 1–11.
- De Bodt, S.; Proost, S.; Vandepoele, K.; Rouzé, P.; Van de Peer, Y. Predicting protein-protein interactions in arabidopsis thaliana through integration of orthology, gene ontology and co-expression. BMC Genom. 2009, 10, 1–15.
- Barnouin, K. Two-dimensional gel electrophoresis for analysis of protein complexes. Protein-Protein Interact. 2004, 261, 479–497.
- Zhang, Y.; Fernie, A.R. On the detection and functional significance of the protein–protein interactions of mitochondrial transport proteins. Biomolecules 2020, 10, 1107.
- Zhang, Y.; Natale, R.; Domingues, A.P.J.; Toleco, M.R.; Siemiatkowska, B.; Fàbregas, N.; Fernie, A.R. Rapid identification of protein-protein interactions in plants. Curr. Protoc. Plant Biol. 2019, 4, e20099.
- Yang, X.; Wen, Z.; Zhang, D.; Li, Z.; Li, D.; Nagalakshmi, U.; Dinesh-Kumar, S.P.; Zhang, Y. Proximity labeling: An emerging tool for probing in planta molecular interactions. Plant Commun. 2020, 100137.
- Mair, A.; Xu, S.-L.; Branon, T.C.; Ting, A.Y.; Bergmann, D.C. Proximity labeling of protein complexes and cell-type-specific organellar proteomes in arabidopsis enabled by turboid. Elife 2019, 8, e47864.
- Liu, X.; Salokas, K.; Weldatsadik, R.G.; Gawriyski, L.; Varjosalo, M. Combined proximity labeling and affinity purification− mass spectrometry workflow for mapping and visualizing protein interaction networks. Nat. Protoc. 2020, 15, 3182–3211.
- Varjosalo, M.; Sacco, R.; Stukalov, A.; Van Drogen, A.; Planyavsky, M.; Hauri, S.; Aebersold, R.; Bennett, K.L.; Colinge, J.; Gstaiger, M. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized ap-ms. Nat. Methods 2013, 10, 307.
- Kim, D.I.; Birendra, K.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461.
- Kwak, C.; Shin, S.; Park, J.-S.; Jung, M.; Nhung, T.T.M.; Kang, M.-G.; Lee, C.; Kwon, T.-H.; Park, S.K.; Mun, J.Y. Contact-id, a tool for profiling organelle contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation. Proc. Natl. Acad. Sci. USA 2020, 117, 12109–12120.
- Bauer, A.; Kuster, B. Affinity purification-mass spectrometry. Eur. J. Biochem. 2003, 270, 570–578.
- Li, Y. The tandem affinity purification technology: An overview. Biotechnol. Lett. 2011, 33, 1487–1499.
- Rohila, J.S.; Chen, M.; Cerny, R.; Fromm, M.E. Improved tandem affinity purification tag and methods for isolation of protein heterocomplexes from plants. Plant J. 2004, 38, 172–181.
- Bontinck, M.; Van Leene, J.; Gadeyne, A.; De Rybel, B.; Eeckhout, D.; Nelissen, H.; De Jaeger, G. Recent trends in plant protein complex analysis in a developmental context. Front. Plant Sci. 2018, 9, 640.
- Wu, S.-C.; Wong, S.-L. Structure-guided design of an engineered streptavidin with reusability to purify streptavidin-binding peptide tagged proteins or biotinylated proteins. PLoS ONE 2013, 8, e69530.
- Née, G.; Tilak, P.; Finkemeier, I. A versatile workflow for the identification of protein–protein interactions using gfp-trap beads and mass spectrometry-based label-free quantification. In Plant Proteomics; Springer: Heidelberg, Germany, 2020; pp. 257–271.
- Besbrugge, N.; Van Leene, J.; Eeckhout, D.; Cannoot, B.; Kulkarni, S.R.; De Winne, N.; Persiau, G.; Van De Slijke, E.; Bontinck, M.; Aesaert, S. Gsyellow, a multifaceted tag for functional protein analysis in monocot and dicot plants. Plant Physiol. 2018, 177, 447–464.
- Schmidt, T.G.; Skerra, A. The strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007, 2, 1528.
- Rubio, V.; Shen, Y.; Saijo, Y.; Liu, Y.; Gusmaroli, G.; Dinesh-Kumar, S.P.; Deng, X.W. An alternative tandem affinity purification strategy applied to arabidopsis protein complex isolation. Plant J. 2005, 41, 767–778.
- Struk, S.; Braem, L.; Walton, A.; De Keyser, A.; Boyer, F.-D.; Persiau, G.; De Jaeger, G.; Gevaert, K.; Goormachtig, S. Quantitative tandem affinity purification, an effective tool to investigate protein complex composition in plant hormone signaling: Strigolactones in the spotlight. Front. Plant Sci. 2018, 9, 528.
- Zhang, Y.; Sampathkumar, A.; Kerber, S.M.; Swart, C.; Hille, C.; Seerangan, K.; Graf, A.; Sweetlove, L.; Fernie, A.R. A moonlighting role for enzymes of glycolysis in the co-localization of mitochondria and chloroplasts. Nat. Commun. 2020, 11, 4509.
- Zhang, Y.J.; Beard, K.F.M.; Swart, C.; Bergmann, S.; Krahnert, I.; Nikoloski, Z.; Graf, A.; Ratcliffe, R.G.; Sweetlove, L.J.; Fernie, A.R.; et al. Protein-protein interactions and metabolite channelling in the plant tricarboxylic acid cycle. Nat. Commun. 2017, 8, 1–11.
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y. The crapome: A contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 2013, 10, 730.
- Nesvizhskii, A.I. Computational and informatics strategies for identification of specific protein interaction partners in affinity purification mass spectrometry experiments. Proteomics 2012, 12, 1639–1655.
- Bürckstümmer, T.; Bennett, K.L.; Preradovic, A.; Schütze, G.; Hantschel, O.; Superti-Furga, G.; Bauch, A. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat. Methods 2006, 3, 1013–1019.
- Kim, D.I.; Roux, K.J. Filling the void: Proximity-based labeling of proteins in living cells. Trends Cell Biol. 2016, 26, 804–817.
- Hesketh, G.G.; Youn, J.-Y.; Samavarchi-Tehrani, P.; Raught, B.; Gingras, A.-C. Parallel exploration of interaction space by bioid and affinity purification coupled to mass spectrometry. In Proteomics; Comai, L.K.J., Mallick, P., Eds.; Springer: New York, NY, USA, 2017; Volume 1550, pp. 115–136.
- Lambert, J.-P.; Tucholska, M.; Go, C.; Knight, J.D.; Gingras, A.-C. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteom. 2015, 118, 81–94.


