 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Miren Altuna | + 1506 word(s) | 1506 | 2021-06-28 10:32:39 | | | |
| 2 | Camila Xu | Meta information modification | 1506 | 2021-07-02 10:56:27 | | |
Video Upload Options
Individuals with Down syndrome (DS) have an increased risk for epilepsy during the whole lifespan, but especially after age 40 years. The increase in the number of individuals with DS living into late middle age due to improved health care is resulting in an increase in epilepsy prevalence in this population. However, these epileptic seizures are probably underdiagnosed and inadequately treated. This late onset epilepsy is linked to the development of symptomatic Alzheimer’s disease (AD), which is the main comorbidity in adults with DS.
1. Introduction
Down syndrome (DS), caused by an extra chromosome 21, is the most frequent cause of intellectual disability of genetic origin [1][2][3][4][5][6]. During the last decades the life expectancy of people of DS has significantly increased [7]. Increased survival coupled with accelerated aging [8] has led to an increase in the prevalence of age-related diseases, most importantly Alzheimer’s disease (AD), which has a cumulative incidence > 90% at age 65 years.
People with DS have a higher prevalence of epilepsy, compared to the general population, similarly to other forms of intellectual disability [1][9][10][11][12]. The first peak/mode of incidence occurs in the early childhood (0–2 years) of life and the second in the 6th decade [13][14][15][16]. Due to its specific clinical presentation and characteristics, it has received a specific name, late onset myoclonic epilepsy in DS (LOMEDS).
We will review the risk factors and clinical characteristics of epilepsy during childhood and adulthood in people with DS, with a special emphasis on LOMEDS.
2. Epilepsy in Early Childhood (0–5 Years)
Similarly to other forms of intellectual disability, children with DS have a higher prevalence of epilepsy than typically developing children [17][18][19]. This risk typically occurs in children before the first year of life, but some seizures such as the febrile seizures (typically developing between 6 months and 5 years) or the Lennox-Gastaut syndrome, can occur until 5–6 years of age. Generalized epilepsies are more frequent than focal epilepsies in the first two decades of life [20].
The increased risk of seizures in children and young adults with DS has been linked to several mechanisms, including frontal and temporal lobe hypoplasia, dyskinesia of dendrites, abnormal neuronal lamination, decreased neuronal density, especially of inhibitory interneurons with a GABAergic inhibitory function, alteration in cell membrane ion channels, specifically of the glutamatergic receptor GluR5 (encoded in chromosome 21), other metabolic disorders associated with the condition of trisomy 21, and to complications derived from hypoxia and/or vascular damage due to a higher prevalence of congenital heart disease and the perioperative risks of its correction [21][20][22][23][24][25][26][27].
West syndrome is characterized by infantile spasms in the context of hypsarrhythmia or a modified hypsarrhythmia pattern in the electroencephalogram. Fortunately, and despite its higher prevalence in DS, the response to early pharmacological treatment with adrenocorticotropic hormone (ACTH) seems to be better than in the general population, with less risk of seizure recurrence in the long term. Complete remission is achieved in more than 90% of cases [20][23][28][29][30][31].
The critical and intercritical EEG patterns vary significantly depending on the type of seizure, and the existence or not of a specific epileptic syndrome, which often are associated with a characteristic EEG pattern. West syndrome, by definition, is associated with the presence of hypsarrhythmia, defined by the presence of continuous discharges of slow waves, spikes or sharp waves, without synchronization between both hemispheres and of high voltage, giving the sensation of an absolute disorder of the electroencephalogram [23][30][31][32].
There are no clinical trials to support the use of one antiepileptic drug over another in the DS population. Therefore, in the absence of specific guidelines, the same assumptions and treatment options are usually made as in the general population. Newer antiepileptic drugs have not proven to be more effective but are generally better tolerated than the classical or first generations drugs.
3. Epilepsy in Children over 5 Years and Young Adulthood
Reliable information about epilepsy with onset in later childhood, young adulthood and middle ages is lacking. First, there is no sufficient information about the evolution of the aforementioned seizures and epileptic syndromes in early childhood. Second, the prevalence estimates of epilepsy in the adolescents and young adults with DS reported in the literature are variable, ranging from 8% to 14% [33]. The most frequent etiologies of epilepsy in this age range are comparable to the general population.
There is a large variability in the type of untriggered seizures in older children and adults with DS between 5 and 40–45 years of age. These epileptic seizures might be focal, with and without altered level of consciousness or generalized motor and non-motor seizures [1][12][34].
The use of routine video-EEG is indicated for the differential diagnosis of episodes that could potentially suggest epileptic etiology. EEG findings must always be interpreted after careful consideration of the clinical context.
If a focal onset is clear, drugs with exclusive indication for focal epilepsy may be used as in the general population. Among the broad-spectrum drugs, the newer drugs have a better safety profile with comparable efficacy and should therefore be recommended.
4. Epileptic Seizures in Late Adulthood (≥40 Years) and Their Relationship with AD
As mentioned before, the second peak in epilepsy incidence is closely associated with the development of symptomatic AD.
The extracellular accumulation of amyloid-β (Aβ) plaques induces synaptic degeneration, circuit remodeling and abnormal synchronization of neuronal networks (Aβ1-42alters Thus potentially favoring increased cortical irritability and inducing a pro-epileptic effect [2][13][35] in individuals who, in addition, have neurodevelopmental structural and cell membrane function abnormalities from birth. The dysfunction of cholinergic and glutamatergic systems (impairment of glial and neuronal uptake of glutamate) in the context of AD impact neuronal excitability and epileptic pathophysiology [36]. Interestingly, soluble Aβ has been associated with decreased GABAergic inhibition and increased hyperexcitability.
The prevalence of untriggered seizures in DS increases dramatically with age after age 45, in parallel with the emergence of symptomatic AD. The prevalence of epilepsy can be as high as 46% in those over 50 years of age, driven by the patients with symptomatic AD. This association is so close that the occurrence of a first episode of an untriggered seizure after the age 40–45 years is highly suggestive of symptomatic AD [12][14][37][35][33][38][39][40].
AD is also an independent risk factor for the development of epilepsy in the general population (prevalence estimates ranging from 10 to 22%) [41][42], and even more so in autosomal dominant AD (prevalence ranging from 19.1 to 47.7%) [43][44]. However, epilepsy is significantly higher in DS, with prevalence estimates ranging from 46 to 84% [12][38]. Thus, the risk is at least 10 times higher in DS than in sporadic AD, and significantly higher than in autosomal dominant AD, including AD patients harboring APP duplications [45][36][14][35][21][43][39][46].
It is important to note that after a single untriggered epileptic seizure in the context of AD in DS antiepileptic treatment should be promptly initiated and maintained.
The most frequent epileptic seizures in the context DSAD are GTCS and myoclonic seizures, which usually coexist. At onset, the seizures have a similar time pattern as in juvenile myoclonic epilepsy, with clear morning predominance upon awakening. They often present in a series of seizures, but the frequency of massive and/or fall-inducing myoclonus is low [45][34][47][21][38][48][49][50][51].
Due to these specific clinical characteristics of this epilepsy associated with symptomatic AD in DS, a specific term has been coined, “late-onset myoclonic epilepsy in DS” (LOMEDS). The clinical course of this myoclonic epilepsy seems to be similar to other progressive myoclonic epilepsies [36][35] (attached figure could be useful to diagnose LOMEDS).
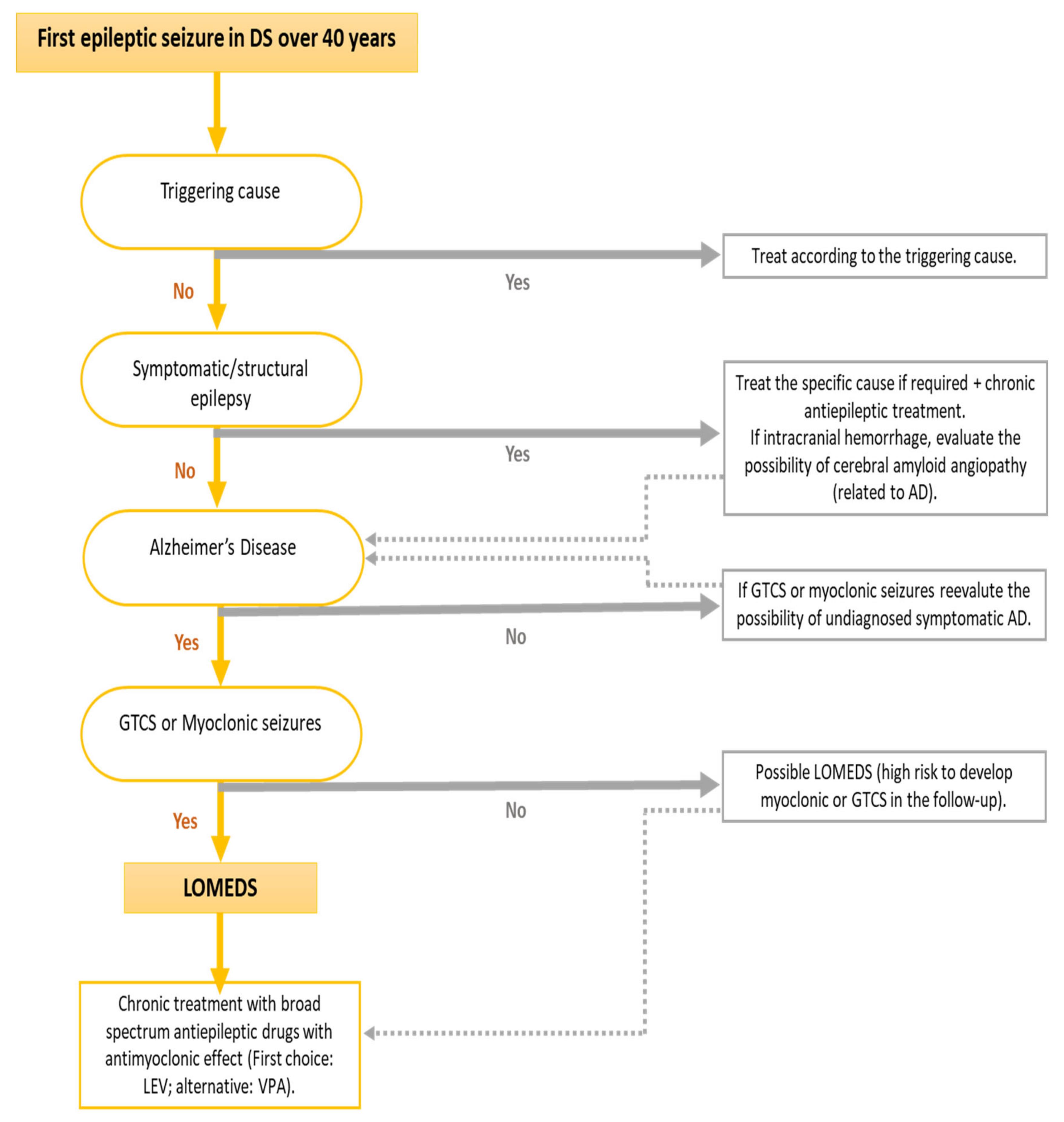
Symptomatic AD usually precedes the onset of the first epileptic seizure, usually by 2–3 years. We note that myoclonic seizures are probably underdiagnosed, which might affect rigorous evaluation of the temporal evolution of seizure onset with respect to that of cognitive impairment due to AD. More advanced AD dementia is associated with a greater risk of developing seizures. [2][35][33][52].
The development of epileptic seizures in DSAD has been associated with a more rapid cognitive and functional decline [36].
As in the general population, the diagnostic performance of the routine video-EEG for the detection of intercritical epileptiform abnormalities is low but increases if it is performed close to occurrence of the seizure, and if performed soon after awakening.
The association between EEG findings and myoclonus supports that these are caused by generalized motor epileptic seizures [53]. This might have therapeutic implications, as myoclonus with a clinical or electroclinical pattern similar to that of juvenile myoclonic epilepsy (JME) might prompt antiepileptic treatment. However, more data is needed to support this guidance.
Good seizure control of both GTCS and myoclonic seizures can be achieved in more than 80% of LOMED cases [2][45][36][54]. The drug of first choice is currently levetiracetam (LEV), which has proven its usefulness in different generalized myoclonic epilepsies [2][35][55][48] and has few drug interactions. It also achieves good control of GTCS and myoclonic seizures [35][21]. On the other hand, it is very important to avoid the use of sodium channel blocking antiepileptic drugs, which have been shown to aggravate myoclonic seizures [36][56].
A personal history of epilepsy, whether LOMEDS or not, does not contraindicate the initiation of acetylcholinesterase inhibitor treatment, nor does it oblige to its suspension (drugs widely used in DSAD) [57]. It is also important to identify and specifically treat comorbidities such as sleep disorders, e.g., OSA, which can worsen seizure control in DSAD.
References
- Kinnear, D.; Morrison, J.; Allan, L.; Henderson, A.; Smiley, E.; Cooper, S.-A. Prevalence of physical conditions and multimorbidity in a cohort of adults with intellectual disabilities with and without Down syndrome: Cross-sectional study. BMJ Open 2018, 8, e018292.
- Lott, I.T.; Doran, E.; Nguyen, V.Q.; Tournay, A.; Movsesyan, N.; Gillen, D.L. Down Syndrome and Dementia: Seizures and Cognitive Decline. J. Alzheimer’s Dis. 2012, 29, 177–185.
- Breia, P.; Mendes, R.; Silvestre, A.; Gonçalves, M.J.; Figueira, M.J.; Bispo, R. Adults with Down syndrome: Characterization of a Portuguese sample. Acta Méd. Port. 2014, 27, 357–363.
- Startin, C.M.; D’Souza, H.; Ball, G.; Hamburg, S.; Hithersay, R.; Hughes, K.M.; Massand, E.; Karmiloff-Smith, A.; Thomas, M.S.; LonDownS Consortium; et al. Health comorbidities and cognitive abilities across the lifespan in Down syndrome. J. Neurodev. Disord. 2020, 12, 4.
- Arvio, M.; Sillanpää, M. Prevalence, aetiology and comorbidity of severe and profound intellectual disability in Finland. J. Intellect. Disabil. Res. 2003, 47 Pt 2, 108–112.
- Tenenbaum, A.; Chavkin, M.; Wexler, I.D.; Korem, M.; Merrick, J. Morbidity and hospitalizations of adults with Down syndrome. Res. Dev. Disabil. 2012, 33, 435–441.
- Rafii, M.S.; Santoro, S.L. Prevalence and Severity of Alzheimer Disease in Individuals with Down Syndrome. JAMA Neurol. 2019, 76, 142.
- Glasson, E.J.; Dye, D.E.; Bittles, A.H. The triple challenges associated with age-related comorbidities in Down syndrome. J. Intellect. Disabil. Res. 2013, 58, 393–398.
- Ohwada, H.; Nakayama, T.; Tomono, Y.; Yamanaka, K. Predictors, including blood, urine, anthropometry, and nutritional indices, of all-cause mortality among institutionalized individuals with intellectual disability. Res. Dev. Disabil. 2013, 34, 650–655.
- Dyggve, H.; Kodahl, T. Disease pattern among 942 mentally retarded persons in a Danish county. Acta Psychiatr. Scand. 1979, 59, 381–394.
- Hermans, H.; Evenhuis, H.M. Multimorbidity in older adults with intellectual disabilities. Res. Dev. Disabil. 2014, 35, 776–783.
- Vignoli, A.; Zambrelli, E.; Chiesa, V.; Savini, M.; La Briola, F.; Gardella, E.; Canevini, M.P. Epilepsy in adult patients with Down syndrome: A clinical-video EEG study. Epileptic Disord. 2011, 13, 125–132.
- McVicker, R.W.; Shanks, O.E.P.; McClelland, R.J. Prevalence and Associated Features of Epilepsy in Adults with Down’s Syndrome. Br. J. Psychiatry 1994, 164, 528–532.
- Lefter, S.; Costello, D.J.; McNamara, B.; Sweeney, B. Clinical and EEG Features of Seizures in Adults with Down Syndrome. J. Clin. Neurophysiol. 2011, 28, 469–473.
- Määttä, T.; Määttä, J.; Tervo-Määttä, T.; Taanila, A.; Kaski, M.; Iivanainen, M. Healthcare and guidelines: A population-based survey of recorded medical problems and health surveillance for people with Down syndrome. J. Intellect. Dev. Disabil. 2011, 36, 118–126.
- Collacott, R.A. Epilepsy, dementia and adaptive behaviour in Down’s syndrome. J. Intellect. Disabil. Res. 2008, 37, 153–160.
- Gaete, B.; Mellado, C.; Hernández, M. Prevalence of neurological disorders among children with Down syndrome. Rev. Med. Chile 2012, 140, 214–218.
- Arya, R.; Kabra, M.; Gulati, S. Epilepsy in children with Down syndrome. Epileptic Disord 2011, 13, 1–7.
- Ulate-Campos, A.; Nascimento, A.; Ortez, C. Down’s syndrome and epilepsy. Int. Med. Rev. Down Syndr. 2014, 18, 3–8. Available online: (accessed on 21 June 2021).
- Gaete, B.; Mellado, C.; Hernández, M. Trastornos neurológicos en niños con síndrome de Down. Rev. Méd. Chile 2012, 140, 214–218.
- De Simone, R.; Daquin, G.; Genton, P. Senile myoclonic epilepsy in Down syndrome: A video and EEG presentation of two cases. Epileptic Disord. 2006, 8, 223–227.
- Shimakawa, S.; Tanabe, T.; Ono, M.; Nonaka, M.; Nambu, M.; Shinohara, T.; Nishikubo, T.; Fukui, M.; Nomura, S.; Azumagawa, K.; et al. Incidence of febrile seizure in patients with Down syndrome. Pediatr. Int. 2015, 57, 670–672.
- Daniels, D.; Knupp, K.; Benke, T.; Wolter-Warmerdam, K.; Moran, M.; Hickey, F. Infantile Spasms in Children with Down Syndrome: Identification and Treatment Response. Glob. Pediatr. Health 2019, 6.
- Araujo, B.; Torres, L.B.; Guilhoto, L.M.F. Cerebal overinhibition could be the basis for the high prevalence of epilepsy in persons with Down syndrome. Epilepsy Behav. 2015, 53, 120–125.
- Takashima, S.; Becker, L.E.; Armstrong, D.L.; Chan, F. Abnormal neuronal development in the visual cortex of the human fetus and infant with Down’s syndrome. A quantitative and qualitative Golgi study. Brain Res. 1981, 225, 1–21.
- Stafstrom, C.E.; Patxot, O.F.; Gilmore, H.E.; Wisniewski, K.E. Seizures in Children with Down Syndrome: Etiology, Characteristics and Outcome. Dev. Med. Child Neurol. 1991, 33, 191–200.
- Gupta, A. Febrile Seizures. Continuum 2016, 22, 51–59.
- Gandy, K.C.; Castillo, H.A.; Ouellette, L.; Castillo, J.; Lupo, P.J.; Jacola, L.M.; Rabin, K.R.; Raghubar, K.P.; Gramatges, M.M. The relationship between chronic health conditions and cognitive deficits in children, adolescents, and young adults with Down syndrome: A systematic review. PLoS ONE 2020, 15, e0239040.
- Kats, D.J.; Roche, K.J.; Skotko, B.G. Epileptic spasms in individuals with Down syndrome: A review of the current literature. Epilepsia Open 2020, 5, 344–353.
- Pueschel, S.M.; Louis, S. Reflex seizures in Down syndrome. Child’s Nerv. Syst. 1993, 9, 23–24.
- Giménez-Roldán, S.; Martin, M. Startle epilepsy complicating down syndrome during adulthood. Ann. Neurol. 1980, 7, 78–80.
- McCarron, M.; Gill, M.; McCallion, P.; Begley, C. Health co-morbidities in ageing persons with Down syndrome and Alzheimer’s dementia. J. Intellect. Disabil. Res. 2005, 49, 560–566.
- Giorgi, F.S.; Saccaro, L.F.; Busceti, C.L.; Biagioni, F.; Fornai, F. Epilepsy and Alzheimer’s Disease: Potential mechanisms for an association. Brain Res. Bull. 2020, 160, 107–120.
- Johannsen, P.; Christensen, J.E.J.; Goldstein, H.; Nielsen, V.K.; Mai, J. Epilepsy in Down syndrome—Prevalence in three age groups. Seizure 1996, 5, 121–125.
- Gholipour, T.; Mitchell, S.; Sarkis, R.A.; Chemali, Z. The clinical and neurobehavioral course of Down syndrome and dementia with or without new-onset epilepsy. Epilepsy Behav. 2017, 68, 11–16.
- Aller-Alvarez, J.; Menéndez-González, M.; Ribacoba-Montero, R.; Salvado, M.; Vega, V.; Suárez-Moro, R.; Sueiras, M.; Toledo, M.; Salas-Puig, J.; Álvarez-Sabin, J. Myoclonic epilepsy in Down syndrome and Alzheimer disease. Neurología 2017, 32, 69–73.
- Inokuchi, R.; Mizuno, N.; Shinohara, K.; Matsubara, T.; Doi, K.; Gunshin, M.; Nakajima, S.; Nakamura, K.; Sato, H.; Ishii, T.; et al. Adult-onset seizures in a patient with Down syndrome and portosystemic shunt. Brain Dev. 2014, 36, 626–629.
- Speller, C.; Johnston, S.; Kanagaratum, S. Epilepsy in Down’s syndrome. Br. J. Psychiatry J. Ment. Sci. 1994, 165, 270.
- Vossel, A.K.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, B.L. Epileptic activity in Alzheimer’s disease: Causes and clinical relevance. Lancet Neurol. 2017, 16, 311–322.
- Lam, A.D.; Sarkis, R.A.; Pellerin, K.R.; Jing, J.; Dworetzky, B.A.; Hoch, D.B.; Jacobs, C.S.; Lee, J.W.; Weisholtz, D.S.; Zepeda, R.; et al. Association of epileptiform abnormalities and seizures in Alzheimer disease. Neurology 2020, 95, e2259–e2270.
- Cortini, F.; Cantoni, C.; Villa, C. Epileptic seizures in autosomal dominant forms of Alzheimer’s disease. Seizure 2018, 61, 4–7.
- Lai, F.; Williams, R.S. A prospective study of Alzheimer disease in Down syndrome. Arch. Neurol. 1989, 46, 849–853.
- Toniolo, S.; Sen, A.; Husain, M. Modulation of Brain Hyperexcitability: Potential New Therapeutic Approaches in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9318.
- Vöglein, J.; Ricard, I.; Noachtar, S.; Kukull, W.A.; Dieterich, M.; Levin, J.; Danek, A. Seizures in Alzheimer’s disease are highly recurrent and associated with a poor disease course. J. Neurol. 2020, 267, 2941–2948.
- D’Orsi, G.; Specchio, L.M. Progressive myoclonus epilepsy in Down syndrome patients with dementia. J. Neurol. 2014, 261, 1584–1597.
- Evenhuis, H.M. The natural history of dementia in Down’s syndrome. Arch. Neurol. 1990, 47, 263–267.
- Crespel, A.; Gonzalez, V.; Coubes, P.; Gelisse, P. Senile myoclonic epilepsy of Genton: Two cases in Down syndrome with dementia and late onset epilepsy. Epilepsy Res. 2007, 77, 165–168.
- Forsgren, L.; Edvinsson, S.O.; Blomquist, H.K.; Heijbel, J.; Sidenvall, R. Epilepsy in a population of mentally retarded children and adults. Epilepsy Res. 1990, 6, 234–248.
- Moller, J.C.; Hamer, H.M.; Oertel, W.H.; Rosenow, F. Late-onset myoclonic epilepsy in Down’s syndrome (LOMEDS). Seizure 2001, 10, 303–306.
- Li, L.M.; O’Donoghue, M.F.; Sander, J.W. Myoclonic epilepsy of late onset in trisomy 21. Arq. Neuropsiquiatr. 1995, 53, 792–794.
- Xu, Y.; Lavrencic, L.; Radford, K.; Booth, A.; Yoshimura, S.; Anstey, K.J.; Anderson, C.S.; Peters, R. Systematic review of coexistent epileptic seizures and Alzheimer’s disease: Incidence and prevalence. J. Am. Geriatr. Soc. 2021.
- McLoughlin, I.J. A Study of Mortality Experiences in a Mental-Handicap Hospital. Br. J. Psychiatry 1988, 153, 645–649.
- Hithersay, R.; Startin, C.M.; Hamburg, S.; Mok, K.Y.; Hardy, J.; Fisher, E.M.C.; Tybulewicz, V.L.J.; Nizetic, D.; Strydom, A. Association of Dementia with Mortality among Adults with Down Syndrome Older than 35 Years. JAMA Neurol. 2019, 76, 152–160.
- Prasher, V. Epilepsy and associated effects on adaptive behaviour in adults with Down syndrome. Seizure 1995, 4, 53–56.
- Arai, Y.; Iwasaki, Y.; Suzuki, T.; Ide, S.; Kaga, M. Elimination of amyloid precursor protein in senile plaques in the brain of a patient with Alzheimer-type dementia and Down syndrome. Brain Dev. 2019, 41, 106–110.
- Giménez, S.; Videla, L.; Romero, S.; Benejam, B.; Clos, S.; Fernández, S.; Martínez, M.; Carmona-Iragui, M.; Antonijoan, R.M.; Mayos, M.; et al. Prevalence of Sleep Disorders in Adults with Down Syndrome: A Comparative Study of Self-Reported, Actigraphic, and Polysomnographic Findings. J. Clin. Sleep Med. 2018, 14, 1725–1733.
- Bubu, O.M.; Andrade, A.G.; Umasabor-Bubu, O.Q.; Hogan, M.M.; Turner, A.D.; de Leon, M.; Ogedegbe, G.; Ayappa, I.; Girardin, J.-L.G.; Jackson, M.L.; et al. Obstructive sleep apnea, cognition and Alzheimer’s disease: A systematic review integrating three decades of multidisciplinary research. Sleep Med. Rev. 2020, 50, 101250.


