 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kohji Fukunaga | + 1235 word(s) | 1235 | 2021-06-18 11:24:39 | | | |
| 2 | Rita Xu | Meta information modification | 1235 | 2021-07-02 06:14:49 | | | | |
| 3 | Conner Chen | Meta information modification | 1235 | 2021-09-22 04:21:33 | | |
Video Upload Options
Lewy bodies are pathological characteristics of Lewy body dementia (LBD) and are composed of α-synuclein (α-Syn), which is mostly degraded via the ubiquitin–proteasome system. More importantly, 26S proteasomal activity decreases in the brain of LBD patients.
1. Introduction
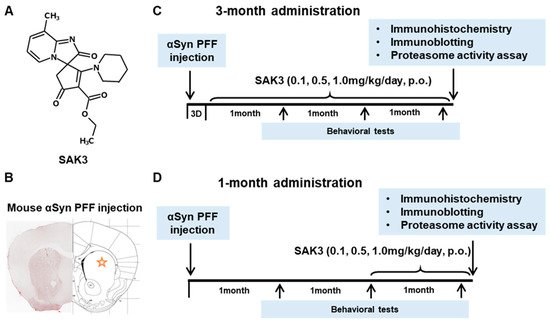
2. SAK3 Prevented the Development of Phosphorylated α-Syn (Ser129) of SNc in PF-Injected Mice
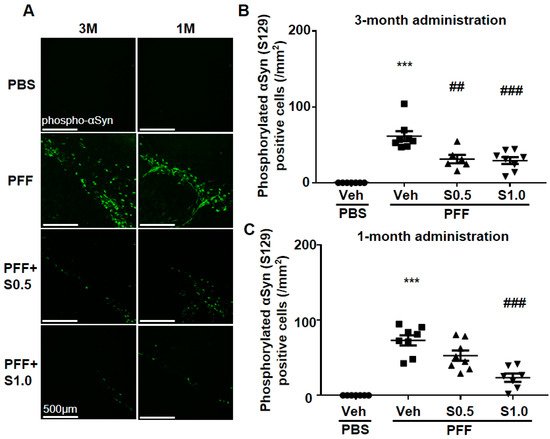
Immunohistochemistry was performed 12 weeks after PFF injection, as described in the schedule shown in Figure 1, and the spread of phosphorylated α-Syn in the substantia nigra pars compacta (SNc) area was analyzed. In the PBS group, phosphorylated α-syn was not detected in any area of the brain. The existing regions of phosphorylated α-Syn in the brain are similar to those reported previously [40]. Phosphorylated α-Syn in PFF-injected mice registered at high levels in the SNc area (3-month administration [3M]: 61.46 ± 6.578, p < 0.0001; 1-month administration [1M]: 73.24 ± 6.726, p < 0.0001; vs. PBS + vehicle; Figure 2A–C), and was decreased by SAK3 administration for 3 months (0.5 mg/kg: 31.24 ± 5.439, p = 0.0017; 1.0 mg/kg: 29.32 ± 4.611, p = 0.0004; vs. PFF + vehicle; Figure 2A,B) and 1 month (1.0 mg/kg: 23.58 ± 5.503, p < 0.0001; vs. PFF + vehicle; Figure 2A,C).
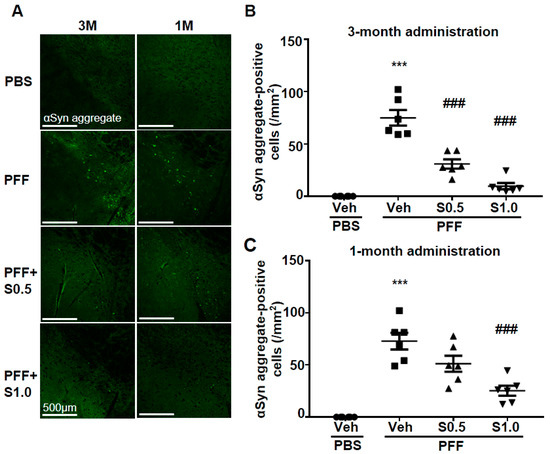
3. SAK3 Attenuated Neuronal Death and α-Syn Aggregation of Dopaminergic Neurons in PFF-Injected Mice
References
- Pillon, B.; Dubois, B.; Cusimano, G.; Bonnet, A.M.; Lhermitte, F.; Agid, Y. Does cognitive impairment in Parkinson’s disease result from non-dopaminergic lesions? J. Neurol. Neurosurg Psychiatry 1989, 52, 201–206.
- Walker, Z.; Possin, K.L.; Boeve, B.F.; Aarsland, D. Lewy body dementias. Lancet 2015, 386, 1683–1697.
- Lippa, C.F.; Duda, J.E.; Grossman, M.; Hurtig, H.I.; Aarsland, D.; Boeve, B.F.; Brooks, D.J.; Dickson, D.W.; Dubois, B.; Emre, M.; et al. DLB and PDD boundary issues: Diagnosis, treatment, molecular pathology, and biomarkers. Neurology 2007, 68, 812–819.
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636.
- Giehm, L.; Svergun, D.I.; Otzen, D.E.; Vestergaard, B. Low-resolution structure of a vesicle disrupting α-synuclein oligomer that accumulates during fibrillation. Proc. Natl. Acad. Sci. USA 2011, 108, 3246–3251.
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3301–3305.
- Brundin, P.; Melki, R. Prying into the Prion Hypothesis for Parkinson’s Disease. J. Neurosci. 2017, 37, 9808–9818.
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953.
- Schampel, A.; Kuerten, S. Danger: High Voltage-The Role of Voltage-Gated Calcium Channels in Central Nervous System Pathology. Cells 2017, 6, 43.
- Jones, E.G. Calcium channels in higher-level brain function. Proc. Natl. Acad. Sci. USA 2007, 104, 17903–17904.
- Blesneac, I.; Chemin, J.; Bidaud, I.; Huc-Brandt, S.; Vandermoere, F.; Lory, P. Phosphorylation of the Cav3.2 T-type calcium channel directly regulates its gating properties. Proc. Natl. Acad. Sci. USA 2015, 112, 13705–13710.
- Powell, K.L.; Cain, S.M.; Snutch, T.P.; O’Brien, T.J. Low threshold T-type calcium channels as targets for novel epilepsy treatments. Br. J. Clin. Pharmacol. 2014, 77, 729–739.
- Perez-Reyes, E.; Cribbs, L.L.; Daud, A.; Lacerda, A.E.; Barclay, J.; Williamson, M.P.; Fox, M.; Rees, M.; Lee, J.H. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature 1998, 391, 896–900.
- Talley, E.M.; Cribbs, L.L.; Lee, J.H.; Daud, A.; Perez-Reyes, E.; Bayliss, D.A. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J. Neurosci. 1999, 19, 1895–1911.
- McCormick, D.A.; Bal, T. Sleep and arousal: Thalamocortical mechanisms. Annu. Rev. Neurosci. 1997, 20, 185–215.
- Lambert, R.C.; Bessaïh, T.; Crunelli, V.; Leresche, N. The many faces of T-type calcium channels. Pflügers Arch. Eur. J. Physiol. 2014, 466, 415–423.
- Nelson, M.T.; Todorovic, S.M.; Perez-Reyes, E. The role of T-type calcium channels in epilepsy and pain. Curr. Pharm. Des. 2006, 12, 2189–2197.
- Yabuki, Y.; Matsuo, K.; Izumi, H.; Haga, H.; Yoshida, T.; Wakamori, M.; Kakei, A.; Sakimura, K.; Fukuda, T.; Fukunaga, K. Pharmacological properties of SAK3, a novel T-type voltage-gated Ca(2+) channel enhancer. Neuropharmacology 2017, 117, 1–13.
- Xu, J.; Yabuki, Y.; Yu, M.; Fukunaga, K. T-type calcium channel enhancer SAK3 produces anti-depressant-like effects by promoting adult hippocampal neurogenesis in olfactory bulbectomized mice. J. Pharmacol. Sci. 2018, 137, 333–341.
- Izumi, H.; Kawahata, I.; Shinoda, Y.; Helmstetter, F.J.; Fukunaga, K. SAK3 Administration Improves Spine Abnormalities and Cognitive Deficits in App(NL-G-F/NL-G-F) Knock-in Mice by Increasing Proteasome Activity through CaMKII/Rpt6 Signaling. Int. J. Mol. Sci. 2020, 21, 3833.
- Palmqvist, S.; Schöll, M.; Strandberg, O.; Mattsson, N.; Stomrud, E.; Zetterberg, H.; Blennow, K.; Landau, S.; Jagust, W.; Hansson, O. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat. Commun. 2017, 8, 1214.
- Bentea, E.; Verbruggen, L.; Massie, A. The Proteasome Inhibition Model of Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 31–63.
- De Vrij, F.M.; Sluijs, J.A.; Gregori, L.; Fischer, D.F.; Hermens, W.T.; Goldgaber, D.; Verhaagen, J.; Van Leeuwen, F.W.; Hol, E.M. Mutant ubiquitin expressed in Alzheimer’s disease causes neuronal death. FASEB J. 2001, 15, 2680–2688.
- Hope, A.D.; de Silva, R.; Fischer, D.F.; Hol, E.M.; van Leeuwen, F.W.; Lees, A.J. Alzheimer’s associated variant ubiquitin causes inhibition of the 26S proteasome and chaperone expression. J. Neurochem. 2003, 86, 394–404.
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194.
- Kawahata, I.; Tokuoka, H.; Parvez, H.; Ichinose, H. Accumulation of phosphorylated tyrosine hydroxylase into insoluble protein aggregates by inhibition of an ubiquitin-proteasome system in PC12D cells. J. Neural Transm. 2009, 116, 1571–1578.
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398.
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428.
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097.
- Pickart, C.M.; Cohen, R.E. Proteasomes and their kin: Proteases in the machine age. Nat. Rev. Mol. Cell Biol. 2004, 5, 177–187.
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 2003, 278, 11753–11759.
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. 2011, 31, 14508–14520.
- Manecka, D.L.; Vanderperre, B.; Fon, E.A.; Durcan, T.M. The Neuroprotective Role of Protein Quality Control in Halting the Development of Alpha-Synuclein Pathology. Front. Mol. Neurosci. 2017, 10, 311.
- McNaught, K.S.; Perl, D.P.; Brownell, A.L.; Olanow, C.W. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann. Neurol. 2004, 56, 149–162.
- Alghamdi, A.; Vallortigara, J.; Howlett, D.R.; Broadstock, M.; Hortobágyi, T.; Ballard, C.G.; Thomas, A.J.; O’Brien, J.; Aarsland, D.; Attems, J.; et al. Reduction of RPT6/S8 (a Proteasome Component) and Proteasome Activity in the Cortex is Associated with Cognitive Impairment in Lewy Body Dementia. J. Alzheimers Dis. 2017, 57, 373–386.
- Dulka, B.N.; Pullins, S.E.; Cullen, P.K.; Moyer, J.R., Jr.; Helmstetter, F.J. Age-related memory deficits are associated with changes in protein degradation in brain regions critical for trace fear conditioning. Neurobiol. Aging 2020, 91, 160–166.
- Giannini, C.; Kloß, A.; Gohlke, S.; Mishto, M.; Nicholson, T.P.; Sheppard, P.W.; Kloetzel, P.M.; Dahlmann, B. Poly-Ub-substrate-degradative activity of 26S proteasome is not impaired in the aging rat brain. PLoS ONE 2013, 8, e64042.
- Jarome, T.J.; Kwapis, J.L.; Ruenzel, W.L.; Helmstetter, F.J. CaMKII, but not protein kinase A, regulates Rpt6 phosphorylation and proteasome activity during the formation of long-term memories. Front. Behav. Neurosci. 2013, 7, 115.
- Hamilton, A.M.; Oh, W.C.; Vega-Ramirez, H.; Stein, I.S.; Hell, J.W.; Patrick, G.N.; Zito, K. Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron 2012, 74, 1023–1030.
- Paumier, K.L.; Luk, K.C.; Manfredsson, F.P.; Kanaan, N.M.; Lipton, J.W.; Collier, T.J.; Steece-Collier, K.; Kemp, C.J.; Celano, S.; Schulz, E.; et al. Intrastriatal injection of preformed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 2015, 82, 185–199.



