Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
Lewy bodies are pathological characteristics of Lewy body dementia (LBD) and are composed of α-synuclein (α-Syn), which is mostly degraded via the ubiquitin–proteasome system. More importantly, 26S proteasomal activity decreases in the brain of LBD patients.
- alpha-synuclein
- Lewy body dementia
- proteasome activity
- Alzheimer’s disease
- amyloid β plaque
- SAK3
1. Introduction
Lewy body dementia (LBD) is the second most common neurodegenerative disease worldwide. In addition to motor dysfunctions such as trembling and slow movements, non-motor dysfunctions, including dementia, depression, and anxiety, are also observed in LBD patients [1]. The main pathological features of LBD are α-synuclein (α-Syn) neuronal inclusions, such as the presence of Lewy bodies (LBs) and neuronal loss [2]; α-Syn is the principal constituent of LBs [3]. There are many forms of α-Syn, such as monomers, oligomers, fibers, and other conformations [4]. Previous reports have suggested that α-Syn oligomers exhibit toxicity in vitro and in vivo [5,6]. A study using a murine model showed that the injection of α-Syn preformed fibrils (PFFs) into the striatum spread to the substantia nigra (SN) [7], similar to prion proteins, leading to the loss of dopamine neurons. Neuronal death and loss of neuronal circuits in the striatum induce cognitive and motor impairments in mice [8]. Therefore, α-Syn plays a crucial role in the pathogenesis and progression of LBD and PD.
T-type voltage-gated calcium channels (T-VGCC), which play a critical role in brain function [9,10], have three electrophysiological characteristics: fast inactivation, slow deactivation kinetics, and low single-channel conductance [11,12]. Three types of T-type calcium channels consisting of Cav3.1, Cav3.2, and Cav3.3 are encoded by CACNA1G, CACNA1H, and CACNA1I genes, respectively, which are expressed in the brain and play essential roles in both physiological and pathological systems, including sleep, pain, and epilepsy [13,14,15,16,17]. Our previous studies confirmed that SAK3 stimulates T-type voltage-gated calcium channels in Cav3.1 and Cav3.3 without affecting Cav3.2 [18]. We also showed that SAK3 improved the impaired cognitive function in olfactory bulbectomized mice via the action of T-VGCC using murine Alzheimer’s disease (AD) models [18,19]. Recently, Izumi et al. reported that SAK3 administration ameliorated cognitive impairments in App NL-F/NL-F(NL-G-F) knock-in mice by improving the synaptic abnormalities in the hippocampal CA1 and cortex. Moreover, they found that SAK3 administration reduced Aβ deposition, which has deleterious effects on the brain, by reducing the proteasome activity [20,21]. The prominent observation in amyloid precursor protein knock-in (APP-KI) mice is that SAK3 administration enhances Ca2+/calmodulin-dependent protein kinase II (CaMKII)/Rpt6 phosphorylation, thereby rescuing the decreased proteasome activity in the APP-KI mouse brain.
The ubiquitin–proteasome system (UPS) is known as an efficient pathway for degrading misfolded or damaged proteins, and it plays a significant role in maintaining the physiological functions of cells [22,23]. UPS dysfunction is often observed in neurodegenerative diseases, such as AD [24] and PD [25,26], with protein misfolding and aggregation. Previous reports have shown that a frameshift mutation in the ubiquitin transcript was explicitly observed in the brains of patients with AD [27,28]. Aβ oligomers can inhibit proteasome activity by directly binding with the 20S proteasome [29]. Previous studies have shown that α-Syn aggregation can directly inhibit the proteolytic active site of the β subunit of the 20S proteasome [30] or bind it to the S6 subunit of the 19S cap [31]. It is uncertain whether protein aggregation leads to proteasomal damage, or vice versa. Under normal physiological conditions, intracellular α-Syn is mostly degraded by the UPS [32,33]. In addition, many pathological symptoms have been observed in PD, such as the formation of Lewy body-like inclusions in rats that received systemic administration of the proteasome inhibitor [34]. However, tissues from clinically diagnosed PD patients were checked and compared with normal aging controls; the expression level and activity of the 20S proteasome decreased significantly in PD nigral neurons that contain α-Syn inclusions [27], and the expression level and activity of the 19S Rpt6 subunit expression was also found in PD patients [35]. In several studies on brain aging, memory deficits with age are associated with reduced phosphorylation of the Rpt6 proteasome regulatory subunit [36]. However, no significant difference in baseline 20S proteasome activity was observed in the hippocampus of young and elderly rats [37].
CaMKII regulates Rpt6 phosphorylation and proteasome activity during the formation of long-term fear memory [38]. Moreover, CaMKII regulates memory formation in the amygdala and phosphorylation of Rpt6, a component of the proteasome 19S subunit [38,39]. Inhibition of CaMKII decreases the rate of new spine growth, and the mutation of Ser120 in the Rpt6 blocks the growth of new spines [38,39]. Considering the SAK3-mediated CaMKII activity, we also investigated the effects of SAK3 on UPS dysfunction in relation to PD.
1. SAK3 Prevented the Development of Phosphorylated α-Syn (Ser129) of SNc in PF-Injected Mice
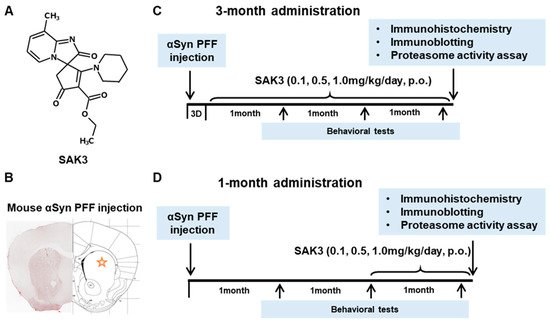
Figure 1. Chemical structure of SAK3, experimental schedule, and α-Synuclein PFF injection area. (A) Chemical structure of SAK3. (B) Position of PFF injection in the mouse brain striatum. Experimental schedule of (C) 3-month chronic administration or (D) 1-month administration of SAK3 in this study.
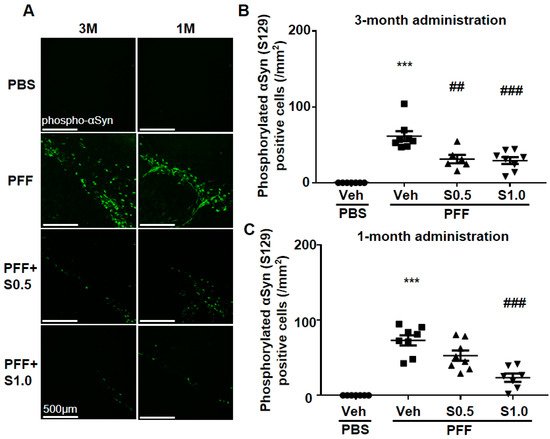
Figure 2. SAK3 chronic administration prevents the spread of phosphorylated α-Syn in PFF-injected mice. (A) Representative immunofluorescence images of phosphorylated α-Syn in the SNc region in both schedules of this study. Scale bar: 500 μm. The number of phosphorylated α-Syn-positive cells was counted in the SNc region for (B) 3 months of the SAK3 treatment schedule (n = 6–8 per group), and (C) 1 month of the SAK3 treatment schedule (n = 7–8 per group). Error bars represent SEM. *** p < 0.001 vs. vehicle-treated PBS injection mice; ## p < 0.01, ### p < 0.001 vs. vehicle-treated α-Syn PFF-injected mice. Abbreviations: Veh = vehicle; S = SAK3.
2. SAK3 Attenuated Neuronal Death and α-Syn Aggregation of Dopaminergic Neurons in PFF-Injected Mice
We checked the level of α-Syn aggregation in dopaminergic neurons. There was no α-Syn aggregation detected in any area of the brain of the PBS-injected mice. On the other hand, in PFF-injected mice, high levels of anti-phosphorylated α-Syn-positive aggregations were observed in the SNc area (3M: 74.97 ± 7.449, p < 0.0001; 1M: 72.81 ± 8.007, p < 0.0001; vs. PBS + vehicle; Figure 3A–C). More importantly, these phosphorylated α-Syn-positive aggregates were decreased by the administration of SAK3 for 3 months (0.5 mg/kg: 30.9 ± 4.414, p < 0.0001; 1.0 mg/kg: 9.67 ± 3.007, p < 0.0001; vs. PFF + vehicle; Figure 3A,B) and for 1 month (1.0 mg/kg: 25.28 ± 4.806, p = 0.0001; vs. PFF + vehicle; Figure 3A,C). These data indicate that SAK3 protects against the formation of phosphorylated α-Syn-positive aggregates in the midbrain.
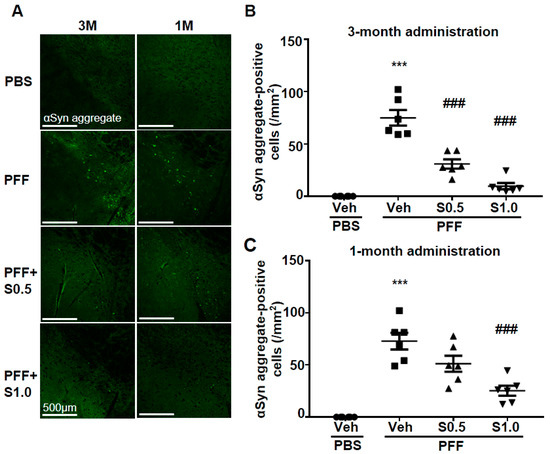
Figure 3. SAK3 treatment prevents the spread of aggregated α-Syn in α-Syn PFF-injected mice. (A) Representative immunofluorescence images of fibrous α-Syn in the SNc region in both schedules of this study. Scale bar: 500 μm. The number of fibrous α-Syn-positive cells was counted in the SNc region for (B) 3 months of the SAK3 treatment schedule (n = 6–8 per group) and (C) 1 month of the SAK3 treatment schedule (n = 7–8 per group). Error bars represent SEM. *** p < 0.001 vs. vehicle-treated PBS-injected mice; ### p < 0.01 vs. vehicle-treated α-Syn PFF-injected mice. Abbreviations: Veh = vehicle; S = SAK3.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22126185
This entry is offline, you can click here to edit this entry!