 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Klaus Grossmann | + 1998 word(s) | 1998 | 2021-07-01 06:02:40 | | | |
| 2 | Lindsay Dong | Meta information modification | 1998 | 2021-07-02 03:37:47 | | |
Video Upload Options
Alzheimer’s disease (AD) is caused by neurodegenerative, but also vascular and hemostatic changes in the brain. The oral thrombin inhibitor dabigatran, which has been used for over a decade in preventing thromboembolism and has a well-known pharmacokinetic, safety and antidote profile, can be an option to treat vascular dysfunction in early AD, a condition known as cerebral amyloid angiopathy (CAA). Dabigatran blocks soluble thrombin, thrombin-mediated formation of fibrin and amyloid-ß protein (Aβ)-containing fibrin clots. These clots are deposited in brain parenchyma and blood vessels in areas of CAA, leading to vascular and blood-brain barrier dysfunction, hypoperfusion and chronically reduced oxygen (hypoxia) and nutrient supply, mainly in hippocampal and neocortical brain areas. Dabigatran has the potential to preserve perfusion and oxygen delivery to the brain, and to prevent parenchymal Aß-, thrombin- and fibrin-triggered inflammatory and neurodegenerative processes, leading to synapse and neuron death, and cognitive decline.
1. Introduction
In Alzheimer’s disease (AD), neurodegenerative but also contributing vascular and hemostatic changes in the brain lead to the loss of memory and cognitive skills, robbing humans ultimately of their essential personality.
Beginning already in the 1970s, clinical studies with small cohorts of patients with senile dementia revealed a beneficial effect on the disease when treated with the anticoagulative vitamin K antagonist (VKA) warfarin [1][2][3]. One study even dates back to the 1960s and found that morbidity and mortality of dementia patients could be reduced by 50% with treatment of the VKA dicumarol [4]. Dicumarol was originally isolated from plant sweet clover hay [5]. It was introduced into clinical medicine in 1941, as the first antithrombotic drug for patients with cardiovascular disease [5]. Recent results on the contribution of cerebrovascular and hemostatic dysfunction to AD have brought the idea back into focus that anticoagulants may also be beneficial for treatment of AD. As key drivers in vascular pathogenesis and derived neurodegenerative changes, progressive accumulation of toxic amyloid-β proteins (Aβ), thrombin and fibrin have recently been identified that can be treated by anticoagulants [6][7].
2. Hemostasis—Anticoagulants Inhibit Thrombin and Fibrin Formation
The multi-stage process of hemostasis prevents excessive blood loss in the organism in the case of injuries to the blood vessel system [8]. In the process of blood clotting (coagulation), a soluble protein from the blood, fibrinogen, is converted into insoluble fibrin, in order to close and heal a wound. Fibrin forms a fiber network with integrated erythrocytes and platelets, a fibrin clot (thrombus). The production of the responsible enzyme, thrombin, which is released from its precursor protein prothrombin, is regulated in a cascade by a variety of tissue and coagulation factors, such as factor Xa. Some of these are activated beforehand in a vitamin K-dependent process [8]. On the other hand, the proteolytic enzyme plasmin degrades fibrin in the fibrinolysis process and thus dissolves fibrin clots [8]. Blood clotting is stimulated and, concomitantly, the risk of formation of harmful thrombi increases, particularly in the case of hereditary modified coagulation factors, large wounds after surgery and injuries, and slower blood flow through incidents, such as atrial fibrillation, damaged blood vessels due to atherosclerosis, and limited physical exercise. These thrombi can trigger occlusion of blood vessels in the process of thrombosis or, after detachment, thrombi move through the vascular system to organs and can cause, e.g., pulmonary embolism or brain infarction [8].
To avoid occurrence of thromboembolism, anticoagulants are used, prophylactically or therapeutically, to inhibit blood clotting.
3. The Role of Thrombin, Fibrin and Amyloid-β-Containing Fibrin Clots
Studies have evidenced that accumulation of toxic Aβ, thrombin and fibrin/fibrinogen co-occur in brains of genetic and sporadic AD patients and in AD mouse brain [9][10][11][12][13][14][15]. It has been found that fibrin(ogen) colonizes together with Aβ along cerebral vessel walls and in the nervous system parenchyma [10][11][15]. Thrombin and fibrin(ogen) trigger neuroinflammatory processes and, furthermore, fibrin(ogen) is able to interact with Aβ [10][11][13][14][15] (Figure 1). In parenchymal tissue, accumulation of fibrin(ogen) and thrombin is caused by the circumstance that BBB is increasingly leaky for plasma proteins, due to the evolving vascular dysfunction in CAA [16][17][18]. Fibrinogen can transgress from the blood vessels into the parenchymal tissue. Here, it is converted into fibrin by thrombin and additional perivascular tissue factor and procoagulant proteins, which are also abundant in this tissue after vascular damage [16][17][18]. Leakage of BBB results from hemorrhage and infarction injuries, and from degeneration of pericytes in capillary walls, which interrupts junctions between adjacent endothelial cells [18]. Overall, BBB dysfunction and resulting processes are associated with increased Aβ production, microglia activation, and malfunctioning and loss of synapses and neurons [19] (Figure 1).
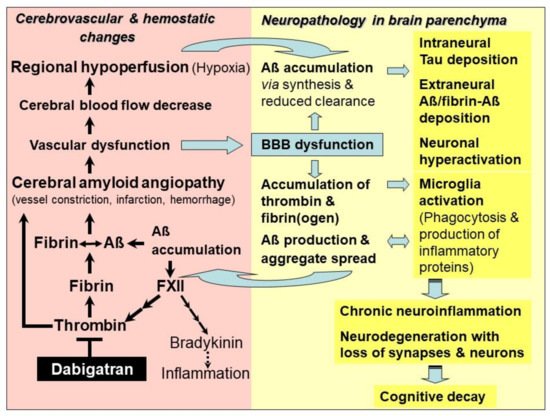
4. Alzheimer’s Disease—Treatment with DOAC-Type Anticoagulants
The new therapeutic approach with DOAC-type anticoagulants relies on their specific mechanism, which acts on key hemostatic drivers in CAA and related neuronal defects in AD. By inhibiting activity or synthesis of thrombin, DOACs are capable of blocking inflammatory thrombin and fibrin for microglia activation, and inhibiting formation of degradation-resistant Aβ-containing fibrin clots. Fibrin-Aβ clots accumulate in the nervous system parenchyma, leading to neuronal damage [11][15]. They also deposit in blood vessels in areas of CAA, where they cause vascular dysfunction, leading to the collapse of BBB, CBF and brain perfusion [9][10]. DOAC treatment has the potential to counteract CAA and vascular dysfunction, and to maintain brain perfusion and supply of oxygen and nutrients. CAA-resulting parenchymal changes, which include accumulation of Aβ, thrombin and fibrin, tauopathies, microglia activation, neuroinflammation, loss of synapses and neurons, and cognitive decline, could also be mitigated or prevented (Figure 1). In order to achieve the best success, AD patients should be treated with DOAC immediately after early diagnosis of vascularly-driven neurodegenerative changes in AD.
5. Alzheimer’s Disease and Other Brain Amyloidosis—Outlook for Therapeutic Use of Dabigatran
DOACs can achieve beneficial effects on vascular and neural dysfunction, contributing to AD. Particularly, the direct thrombin inhibitor dabigatran opens up the opportunity to effectively combat both the accumulation and harmful effect of inflammatory thrombin and fibrin, as well as the production and deposition of Aβ and fibrin-Aβ clots in cerebral blood vessels and parenchyma. Consequently, dabigatran could be able to treat CAA and related vascular dysfunction, in order to mitigate or prevent resulting effects, such as BBB dysfunction, CBF decrease, brain hypoperfusion and lack of oxygen and nutrients, as well as progressive neuroinflammatory, degenerative and cognitive changes (Figure 1).
In conclusion, there are strong rationales that would warrant realization of a first pilot clinical study using dabigatran against AD [6][7][9][13]. These rationales result from (1) recent research on the roles of thrombin, fibrin and Aβ in triggering vascular and neurological changes in AD [9][10][13][17], (2) preclinical studies showing beneficial effects of dabigatran treatments [20][21][22][23] and (3) clinical observer studies [24][25][26][27]. For such a clinical pilot study, the oral direct thrombin inhibitor dabigatran with its well-known application and safety profile, including availability of a fast-acting antidote [28][29], would be a reasonable drug solution to investigate the therapeutic value of thrombin inhibition in AD. This intervention study should ideally be carried out in close cooperation between neurological and cardiovascular medicine. In this way, dabigatran effects on the early course of CAA and neuropathy in AD patients could be studied comprehensively and competently. The possible method repertoire used could include position emission tomography (PET) and magnetic resonance imaging (MRI) technologies for visualizing brain biomarkers, such as Aβ deposits, bleeding events, and CBF dynamics [30][31][32][33]. Invasive cerebrospinal-fluid (CSF) analyses, e.g., on Aβ, tau [33], and preferentially, blood biomarker tests, which indicate neurodegenerative (e.g., Nfl [34], Aβ fragments [33]) and hemostatic alterations (e.g., thrombin, fibrin clot forming), as well as cognitive ability checks, can supplement the study. However, cost/benefit of the intended methods should be parsed before the analysis [35]. Highly important for this clinical study is that participating persons are carefully evaluated, particularly according to their bleeding risk, using, e.g., clinical HAS-BLED-Score criteria and imaging methods [7][8]. As with any drug, benefit as well as risk of undesirable effects must be individually weighted and discussed, as the basis for consent of participants. It is also extremely critical for the success of the therapy with dabigatran that subjects, who are at the beginning of AD pathogenesis, are selected according to a lesson learned from past clinical studies on Aβ-targeting therapies [35]. At best, the onset of AD should be diagnosed very early, using, e.g., AD blood biomarkers and confirmation by CSF analysis and brain imaging methods, if cognitive failures are already suspected or feared. Then, after positive diagnosis, treatment with dabigatran should start immediately, in order to possibly combine the highest therapeutical efficiency with the lowest risk of bleeding. In the case of individuals with genetically high AD risk predisposition (early onset AD), dabigatran could/should be administered prophylactically, before this terrible disease begins.
References
- Ratner, J.; Rosenberg, G.; Kral, V.A.; Engelsmann, F. Anticoagulant therapy for senile dementia. J. Am. Geriatr. Soc. 1972, 20, 556–559.
- Walsh, A.C.; Walsh, B.H.; Melaney, C. Senile-presenile dementia: Follow-up data on an effective psychotherapy-anticoagulant regimen. J. Am. Geriatr. Soc. 1978, 26, 467–470.
- Barber, M.; Tait, C.; Scott, J.; Rumley, A.; Lowe, D.O.; Stott, D.J. Dementia in subjects with atrial fibrillation: Hemostatic function and the role of anticoagulation. J. Throm. Haemost. 2004, 2, 1873–1878.
- Whittier, J.R.; Korenyi, C.; Klein, D.F.; Foley, W. Prevention of degenerative disease: A controlled study of anticoagulant prophylaxis. J. Chronic Dis. 1961, 14, 203–212.
- Kresge, N.; Simoni, R.D.; Hill, R.L. Hemorrhagic sweet clover disease, dicumarol, and warfarin: The work of Karl Paul Link. J. Biolog. Chem. 2005, 280, e5–e6.
- Grossmann, K. Anticoagulants for treatment of Alzheimer’s disease. J. Alzheimers Dis. 2020, 77, 1373–1382.
- Grossmann, K. Direct oral anticoagulants: A new therapy against Alzheimer’s disease? Neural. Reg. Res. 2021, 16, 1556–1557.
- Grosser, T.; Weber, A.-A. Pharmakologie der Hämostase. In Allgemeine und Spezielle Pharmakologie und Toxikologie, 12th ed.; Aktories, K., Förstermann, U., Hofmann, F., Starke, K., Eds.; Elsevier: München, Germany, 2017; pp. 465–488.
- Cortes-Canteli, M.; Iadecola, C. Alzheimer’s disease and vascular aging. J. Am. Coll. Cardiol. 2020, 75, 942–951.
- Strickland, S. Blood will out: Vascular contributions to Alzheimer’s disease. J. Clin. Investig. 2018, 128, 556–563.
- Cortes-Canteli, M.; Paul, J.; Norris, E.H.; Bronstein, R.; Ahn, H.J.; Zamolodchikov, D.; Bhuvanendran, S.; Fenz, K.M.; Strickland, S. Fibrinogen and β-amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer’s disease. Neuron 2010, 66, 695–709.
- Ahn, H.J.; Zamolodchikov, D.; Cortes-Canteli, M.; Norris, E.H.; Glickman, J.F.; Strickland, S. Alzheimer’s disease peptide β-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. USA 2010, 107, 21812–21817.
- Iannucci, J.; Renehan, W.; Grammas, P. Thrombin, a mediator of coagulation, inflammation, and neurotoxicity at the neurovascular interface: Implications for Alzheimer’s disease. Front. Neurosci. 2020, 14, 762.
- Zamolodchikov, D.; Berk-Rauch, H.E.; Oren, D.A.; Stor, D.S.; Singh, P.K.; Kawasaki, M.; Aso, K.; Strickland, S.; Ahn, H.J. Biochemical and structural analysis of the interaction between β-amyloid and fibrinogen. Blood 2016, 128, 1144–1151.
- Cortes-Canteli, M.; Mattei, L.; Richards, A.T.; Norris, E.H.; Strickland, S. Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol. Aging 2015, 36, 608–617.
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201.
- Petersen, M.A.; Ryu, J.K.; Akassoglou, K. Fibrinogen in neurological diseases: Mechanisms, imaging and therapeutics. Nat. Rev. 2018, 19, 283–301.
- Profaci, C.P.; Munij, R.N.; Pulido, R.S.; Daneman, R. The blood-brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, e20190062.
- Deane, R.; Wu, Z.; Zlokovic, B.V. RAGE (yin) versus LRP (yang) balance regulates Alzheimer’s amyloid β-peptide clearance through transport across the blood-brain barrier. Stroke 2004, 35 (Suppl. I), 2628–2631.
- Tripathy, D.; Sanchez, A.; Yin, X.; Luo, J.; Martinez, J.; Grammas, P. Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front. Aging Neurosci. 2013, 5, 19.
- Marangoni, M.N.; Braun, D.; Situ, A.; Moyano, A.L.; Kalinin, S.; Polak, P.; Givogri, M.; Feinstein, D.L. Differential effects on glial activation by a direct versus indirect thrombin inhibitor. J. Neuroimmunol. 2016, 297, 159–168.
- Cortes-Canteli, M.; Kruyer, A.; Fernandez-Nueda, I.; Marcos-Diaz, A.; Ceron, C.; Richards, A.T.; Jno-Charles, O.C.; Rodriguez, I.; Callejas, S.; Norris, E.; et al. Long-term dabigatran treatment delays Alzheimer’s disease pathogenesis in the TgCRND8 mouse model. J. Am. Coll. Cardiol. 2019, 74, 1910–1923.
- Bogatkevich, G.S.; Ludwicka-Bradley, A.; Nietert, P.J.; Akter, T.; van Ryn, J.; Silver, R.M. Antiinflammatory and antifibrotic effects of the oral direct thrombin inhibitor dabigatran etexilate in a murine model of interstitial lung disease. Arthritis Rheum. 2011, 63, 1416–1425.
- Friberg, L.; Rosenqvist, M. Less dementia with oral anticoagulation in atrial fibrillation. Eur. Heart J. 2018, 39, 453–460.
- Mongkhon, P.; Fanning, L.; Lau, W.C.Y.; Tse, G.; Lau, K.K.; Wei, L.; Kongkaew, C.; Wong, I.C.K. Oral anticoagulant and reduced risk of dementia in patients with atrial fibrillation: A population-based cohort study. Heart Rhythm 2020, 17, 706–713.
- Jacobs, V.; May, H.T.; Bair, T.L.; Crandall, B.G.; Cutler, M.J.; Day, J.D.; Mallender, C.; Osborn, J.S.; Stevens, S.M.; Weiss, J.P.; et al. Long-term population-based cerebral ischemic event and cognitive outcomes of direct oral anticoagulants compared with warfarin among long-term anticoagulated patients for atrial fibrillation. Am. J. Cardiol. 2016, 118, 210–214.
- Cheng, W.; Liu, W.; Li, B.; Li, D. Relationship of anticoagulant therapy with cognitive impairment among patients with atrial fibrillation. A meta-analysis and systemic review. J. Cardiovasc. Pharmocol. 2018, 71, 380–387.
- Van Ryn, J.; Goss, A.; Hauel, N.; Wienen, W.; Priepke, H.; Nar, H.; Clemens, A. The discovery of Dabigatran etexilate. Front Pharmacol. 2013, 4, 12.
- Pollack, C.V.; Paul, M.D., Jr.; Eikelboom, J.; Glund, S.; Verhamme, P.; Bernstein, R.A.; Dubiel, R.; Hulsman, M.V.; Hylek, E.M.; Kamphuisen, P.W.; et al. Idarucizumab for dabigatran reversal. N. Engl. J. Med. 2015, 373, 511–520.
- Maier, F.C.; Wehrl, H.F.; Schmid, A.M.; Mannheim, J.G.; Wiehr, S.; Lerdkrait, C.; Calaminus, C.; Stahlschmidt, A.; Ye, L.; Burnet, M.; et al. Longitudinal PET-MRI reveals β-amyloid deposition and rCBF dynamics and connects vascular amyloidosis to quantitative loss of perfusion. Nat. Med. 2014, 20, 1485–1492.
- DeSimone, C.V.; Graff-Radford, J.; El-Harasis, M.A.; Rabinstein, A.A.; Asirvatham, S.J.; Holmes, D.R., Jr. Cerebral amyloid angiopathy: Diagnosis, clinical implications, and management strategies in atrial fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1173–1182.
- Dai, W.; Lopez, O.L.; Carmichael, O.T.; Becker, J.T.; Kuller, L.H.; Gach, H.M. Mild cognitive impairment and Alzheimer’s disease: Patterns of altered cerebral blood flow at MR imaging. Radiology 2009, 250, 856–866.
- Dolgin, E. A tough spot. Nature 2018, 559, S10–S12.
- Preische, O.; Schultz, S.A.; Apel, A.; Kuhle, J.; Kaeser, S.A.; Barro, C.; Gräber, S.; Kuder-Buletta, E.; LaFougere, C.; Laske, C.; et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med. 2019, 25, 277–283.
- McDade, E.; Bateman, R.J. Stop Alzheimer’s before it starts. Nature 2017, 547, 153–155.


