Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | chaobo chen | + 2397 word(s) | 2397 | 2021-05-13 08:12:27 | | | |
| 2 | Catherine Yang | Meta information modification | 2397 | 2021-06-30 04:27:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chen, C. Cholestatic Liver Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/11476 (accessed on 08 February 2026).
Chen C. Cholestatic Liver Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/11476. Accessed February 08, 2026.
Chen, Chaobo. "Cholestatic Liver Disease" Encyclopedia, https://encyclopedia.pub/entry/11476 (accessed February 08, 2026).
Chen, C. (2021, June 29). Cholestatic Liver Disease. In Encyclopedia. https://encyclopedia.pub/entry/11476
Chen, Chaobo. "Cholestatic Liver Disease." Encyclopedia. Web. 29 June, 2021.
Copy Citation
Cholestatic liver diseases including primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) are associated with active hepatic fibrogenesis, which can ultimately lead to the development of cirrhosis.
cholangiocytes
hepatic stellate cells (HSCs)
periductular fibroblasts
1. Introduction
Cholestasis is a chronic liver disease characterised by bile flow obstruction in the liver, bile acid (BA) accumulation, and increased BA concentration in the systemic circulation. Thus, during cholestasis impaired bile formation and processing with insufficient bile reaching the duodenum, leads to the accumulation of intrahepatic and systemic BAs and other potentially toxic cholephilic bacteria. The aetiology of cholestasis includes disorders of bile secretion by hepatocytes and/or biliary epithelial cells (BECs), mechanical processes (e.g., stones, tumours) destroying/blocking smaller and/or larger intrahepatic bile ducts, or immune-mediated fibrotic cholangitis, such as primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) [1][2]. Therefore, BA metabolism plays an important role since the obstruction of bile flow leads to cholestatic injuries, such as in PBC and PSC [3].
Therapies for these diseases are, however, limited. Although ursodeoxycholic acid (UDCA) treatment can significantly improve the prognosis of PBC patients and prolong transplant-free survival, the treatment options for those who do not respond to UDCA remain scarce. Other drugs associated with novel therapies in cholestatic diseases (PBC/PSC) are still in clinical trials, including obeticholic acid (OCA) [4], all-trans retinoic acid (ATRA) [5], Bezafibrate [6], Fenofibrate [7], etc. [8][9]. In addition, there are currently no available drugs to treat PSC [10]. Many of these disorders become chronic, therefore leading eventually to biliary cirrhosis and the need for liver transplantation [11]. Cholestatic liver disease can also cause liver failure and increase the risk of hepatocellular carcinoma (HCC) or cholangiocarcinoma (CCA) [12][13].
2. Primary Biliary Cholangitis (PBC)
PBC, formerly known as primary biliary cirrhosis, is acquired chronic cholestasis related to the autoimmune destruction of small bile ducts, causing portal vein infiltration and fibrosis. PBC is a chronic progressive disease that leads to end-stage liver disease and its related complications [14][15][16]. It can progress to biliary cirrhosis, portal hypertension, liver failure, and is associated with esophagogastric variceal bleeding, ascites, and hepatic encephalopathy [17]. PBC is a destructive lymphocytic cholangitis and specific antimitochondrial antibodies (AMAs) target specific mitochondrial autoantigens [18]. It is characterised by the progressive damage and destruction of biliary epithelial cells (BECs; also called cholangiocytes) and increased portal vein inflammation and fibrosis [19][20], with chronic histological evidence, nonsuppurative, granulomatous, and lymphocytic cholangitis [17]. Simultaneously, in association with PBC, there are also symptoms that markedly affect the quality of life, including cholestatic pruritus, Sjogren’s syndrome, abdominal discomfort, and fatigue [21][22].
Approximately 95% of PBC patients are middle-aged women [23]. Interestingly, this disease rarely affects children [24]. The reports vary worldwide from 1970 to 2014, the annual incidence ranges from 0.3 to 5.8 per 100,000, and the prevalence rates range from 1.9 to 40.2 per 100,000 individuals, respectively, due to increased incidence and improved survival [25][26][27]. From 2004 to 2014, in the United States, the prevalence of PBC increased significantly from 21.7 to 39.2 per 100,000, of which women rose from 33.5 to 57.8 per 100,000 (an increase of 72%), while the incidence rate in men increased from 7.2 to 15.4 per 100,000 (an increase of 114%) [28].
Risk factors for PBC include genetic factors such as the human leukocyte antigen (HLA) and non-HLA allelic variants) [29][30], as well as environmental stimuli. In addition to the regional differences in disease prevalence and family risk, the relationship between epidemiology and bacterial infection, xenobiotics, and smoking history also emphasises the importance of environmental triggers in the pathogenesis of PBC [31][32][33][34][35][36][37]. Furthermore, PBC development has also been linked to microRNA [38] and epigenetic regulation [18]. Moreover, the gut–liver axis is also involved in PBC development. Intestinal dysbacteriosis can affect the bile acid pool and regulate bile acid-activated receptors, which disturbs bile acid metabolism [39][40]. Simultaneously, several lines of evidence suggested that dysbiosis of gut microbiota can destroy the immune homeostasis, thus promoting PBC [41][42].
Population-based historical data from the UK show that about 25% of untreated “classic PBC” patients develop chronic liver failure during this period [43]. An early prospective study found that more than 50% of patients with stage I-III PBC developed histologically confirmed cirrhosis within four years [44]. As cirrhotic individuals, PBC patients may develop complications due to the chronic nature of the disease. The presence of cirrhosis, regardless of its aetiology, is a major risk factor for hepatocellular carcinoma (HCC) or cholangiocarcinoma (CCA) [13][45].
3. Primary Sclerosing Cholangitis (PSC)
PSC is associated with liver damage, characterised by intrahepatic or extrahepatic bile duct injury, and fibrosis of the bile ducts inside and outside the liver, resulting in strictures of the bile ducts and obstruction of bile flow. Clinical manifestations reflect the potential sequence of bile duct injury and fibrosis leading to stricture, cholestasis, and biliary cirrhosis with progressive liver dysfunction [46]. PSC is a male-dominant disease when it is associated with inflammatory bowel disease (IBD), (65–70%), with a male-female ratio of approximately 2:1 [23][47][48][49]. Epidemiological studies show that the prevalence of PSC is about 1/10,000 cases globally, while the incidence rate in northern Europe and the United States is 0.4/100,000 to 2.0/100,000 per year [50][51]. Simultaneously, the survival rate of PSC is increasing [47][52][53][54], which may be partly attributed to early diagnosis due to the application of magnetic resonance cholangiography (MRC). The clinical characteristics of newly diagnosed patients remain stable over time, while no new diagnostic methods were introduced during this period such as before fibrosis occurs [47].
PSC is a typical complex disease with genetic and environmental risk factors. This important genome-wide association has shown that PSC risk is associated with certain phenotypes of human leukocyte antigens (HLA), particularly HLA-DR6, HLA-DR3, and HLA-B8, suggesting the presence of autoimmune disorders in patients with PSC [55]. The risk of PSC is also associated with, at least, 23 regions of the genome [56]. At present, no clear causal environmental factor has been identified, however, the geographical distribution of the disease in northern Europe provides some clues to consider the source of environmental risk factors [57]. Indeed, differences in lifestyle, diet, and living conditions are highly regional [46]. Clinically, inflammatory bowel disease (IBD) is the strongest condition associated with PSC—approximately 70% of patients with PSC having also IBD [58][59][60].
Bacterial cholangitis, osteoporosis, liver cirrhosis, and IBD can be caused by the progression of PSC [61][62][63][64][65][66][67]. It can lead to colorectal neoplasia, pancreatic cancer, CCA, and gallbladder carcinoma [58][65][66][68][69][70]. Large population-based studies indicate that the risk of death in patients with PSC is increased fourfold, in comparison with the general population [47]. The most common causes of death associated with PSC are CCA (32%), liver failure (15%), transplant-related complications (9%), and colorectal cancer (8%), demonstrating that the increased risk of malignancy for PSC has a significant impact on life expectancy [71].
4. Signalling Pathways Involved in Pathogenesis
After cholangiocyte injury, infiltration of immune cells occurs, activating Notch, hedgehog (Hh), Wnt, and other signalling pathways—inducing the increase in fibroblasts due to HSCs activation, which further promote BECs proliferation. Subsequent pathological sequelae including biliary stricture formation, bile retention, bile-related toxic stress, inflammatory cells, or formation of immune cells around the bile duct further aggravate the disease.
4.1. Notch Signalling Pathway
In the past few years, the mechanisms and influence of Notch signalling in liver fibrosis have been developed. Rat HSCs express the Notch receptor in vitro and begin to express JAG1 after activation and differentiation into myofibroblast-like cells [72]. The expression of Notch 2/3, Hey 1/2 increases significantly in the process leading from quiescent HSCs into activated myofibroblasts [73]. High activation of the Notch signal was also observed in hepatic progenitor cells isolated from tissues with PBC [74]. In addition, the number of Notch 1/3/4-positive cells increased significantly in the fibrotic area of Chemokine (C-C motif) ligands 4 (CCl4)-injured rats [75]. In the CCl4-induced rat liver fibrosis model, Notch signalling was also hyperactivated [76]. Moreover, inhibition of Notch in CCl4-induced liver injury significantly damaged HSC activation and triggered the development of fibrosis, and inhibition of this pathway in the liver can prevent or ameliorate fibrosis [77].
During liver fibrosis development, Notch interacts with other signalling pathways, such as TGF-β, Hippo, and Hh, whilst crosstalk between TGF-β activation and Notch occurs in liver fibrosis [78][79] (Figure 1). TGF-β in the liver may partly promote fibrosis by stimulating Notch activity in HSCs. KCs together with bone marrow-derived macrophages are thought to be the main source of TGF-β1 thus promoting the development of liver fibrosis [80][81]. Recent studies have shown that TGF-β2-induced expression of fibrosis genes in cholangiocytes and HSCs is related to the specific regulation of the Notch3 signalling pathway [82]. SOX9, a transdifferentiated biomarker of BECs specifically expressed in cholangiocytes, is also a downstream target of Notch signalling. After BDL in rats, Notch receptor activation, combined with overexpression of SOX9, enhanced BEC proliferation and induced hyper-hepatic fibrosis [83]. Recently, another study from Athwal et al. [84] demonstrated the relationship between increased SOX9 and activation of the Hippo pathway in the development of liver fibrosis. Inhibition of YAP1 in CCl4 and BDL-induced liver fibrosis by injection of specific YAP1-related inhibitor Verteporfin resulted in decreased expression of SOX9 in HSCs. Notch and YAP1 may activate SOX9 in different cell types, both leading to HSCs activation and fibrosis induction [84][85][86]. Noticeably, YAP1/Hippo pathways are related to the activation and amplification of ductular reactive cells (DRC) [87][88]. Additionally, during tissue repair, which normally requires cell proliferation, the Hippo pathway is often downregulated by phosphorylation of its main effectors, yes-associated protein 1 (YAP1) and transcriptional co-activator with PDZ-binding motif (TAZ) [89]. In particular, macrophage-derived TNF-related weak inducer of apoptosis kinase (TWEAK) induced expansion of progenitor cells and proliferation of bile ducts in healthy mice, while fibroblast growth factor–inducible 14 (Fn14) deficient mice or neutralisation of TWEAK prevented the expansion of progenitor cells in cholestasis mice [90][91].
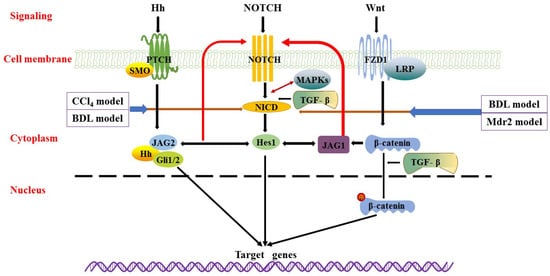
Figure 1. Wnt/β-catenin, Notch, and Hedgehog signalling pathways. Wnt/β-catenin signalling is transmitted through the Frizzled (FZD) receptor, thereby stabilizing β-catenin. Phosphorylated β-catenin translocated into the nucleus to regulate the expression of target genes. There is crosstalk between Wnt/β-catenin signalling and Notch/TGF-β signalling. In Notch signalling, binding of Notch ligands to the receptor results in two proteolytic cleavages to release NICD. The released NICD then translocated into the nucleus, activating transcription factors Hes1, JAG1, and JAG2, whilst the Notch signal pathway interacts with Hh and Wnt signalling pathways. In Hedgehog signalling, Hh ligand secreted by Hedgehog secretory cells binds to PTCH or SMO and generates activated Gli that translocated to the nucleus, inducing target gene expression. These main survival pathways and sophisticated interactions between signalling pathways (i.e., TGF-β and MAPKs) constitute a complex regulatory network for the survival and proliferation of BECs.
4.2. Hedgehog (Hh) Signalling Pathway
Hh is one of the morphogenetic signalling pathways. There are three mammalian Hh proteins—Sonic hedgehog (Shh), Indian Hedgehog (Ihh), and Desert Hedgehog (Dhh). The downstream effector of Hh signalling is Gli1/2, a transcription repressor of the polycomb group and a central regulator of normal stem cell self-renewal. Regardless of the aetiology, it has been proved that the Hh signal is upregulated in the injured liver [92], and it increases with worsening of liver injury and fibrosis [93]. A previous study demonstrated liver accumulation of Hh ligands and activation of the Hh signalling pathway in the livers of BDL rodents and PBC patients [94]. Hh and Notch stimulate each other to promote HSCs activation and subsequent fibrosis [95]. In cholangiocytes, Notch promotes Shh signal transduction by regulating transport inside and outside primary cilium (PC), while Shh promotes Notch signal transduction by directly upregulating Hairy and enhancer of split-1 (Hes1) and Jagged canonical Notch ligand 2 (JAG2) [96] (Figure 2). In vitro and in vivo studies have shown that the Notch signalling pathway drives epithelial to mesenchymal transition (EMT). The interaction between Notch and Hh signalling pathways promotes the transdifferentiation of HSCs into myofibroblasts that involves an EMT, triggering fibrogenesis [76][95]. Finally, Notch also enhances the inflammatory response and M1 polarisation of macrophages [97].
4.3. Wnt Signalling Pathway
The Wnt signalling pathway is likely one of the main factors contributing to the progression of cholestatic liver disease. Wnt signal transduction induces liver fibrosis by promoting HSCs proliferation and activation, accompanied by ECM synthesis, EMT increase, or interaction with other fibrosis mediators [98][99]. Moreover, excessive accumulation of ECM is considered to be a key event in the pathogenesis of ageing-related liver fibrosis [100][101]. Several studies have shown that Wnt signalling is involved in the progression of liver fibrosis, and many components are upregulated and implicated in this process [102][103][104][105] (Figure 1). Conversely, the expression of some Wnt receptors (such as Frizzled 1 (FZD1)) and co-receptor low-density lipoprotein receptor-related protein5/6 (LRP5/6) in activation of HSCs increased in the progression of liver fibrosis but with decreased expression of FZD4/8 [105][106][107][108]. Furthermore, β-catenin is the main downstream effector of classical Wnt signalling, and the loss function of β-catenin will affect the metabolism of BAs. In the BDL model, complete obstruction of bile flow can lead to hepatic cholestasis. Therefore, the enhanced inhibition of BAs synthesis by inhibiting or β-catenin loss can improve the progression of cholestasis. Interestingly, Pradhan [109] showed that the knockdown of β-catenin in Mdr2−/− mice resulted in increased inflammation, cell senescence, promoted fibrosis, and impaired liver regeneration after injury. In Mdr2−/− mice, this phenotype is mainly driven by the toxicity of BAs lacking phospholipids. Therefore, reducing injury, improving regenerative response, and/or maintaining bile flow to prevent stasis are essential for maintaining the liver function, at least in mice.
4.4. Other Related Signalling Pathways
In addition to the above signalling pathways, other studies have found that the c-Jun N-terminal kinase (JNK), a member of the MAPKs family, is involved in the regulation of proliferation, cell death, inflammation, and metabolism [110][111]. JNK contributes to the activation of HSCs, induces overexpression of αSMA during the procession of liver fibrosis [112][113], and promotes the production of myofibroblasts. Kluwe et al. [113] showed that the phosphorylation of JNK increased significantly in mouse liver after BDL or CCl4 administration, as well as in the human fibrotic liver, mainly in fibroblasts. In vivo, the inhibition using a pan-JNK inhibitor did not affect liver injury but significantly reduced fibrosis after BDL or CCl4. JNK1-deficient mice showed reduced fibrosis after BDL or CCl4, while JNK2-deficient mice showed increased fibrosis after BDL but no change after CCl4. In culture, pan-JNK inhibitors prevent the activation of human HSCs induced by TGF-β, PDGF, and angiotensin II-induced murine HSCs activation, and reduce PDGF and TGF-β signal transduction. Zhao [112] showed the specific role of Jnk1 in HSC activation and ECM formation. The absence of Jnk1 correlated with a lower proliferation and survival of HSCs, demonstrating the pivotal contribution of Jnk1 in the development of liver fibrosis in HSCs. However, signalling pathways involved in JNK activation related to liver fibrosis are also participate in bidirectional crosstalk, including TNF-α and NF-κB [114]. Collectively, sustained activation of pre-fibrosis-related signalling pathways may promote the progression of liver fibrosis, combined with immune cell infiltration around biliary tracts.
References
- Erlinger, S. What is cholestasis in 1985? J. Hepatol. 1985, 1, 687–693.
- Fuchs, C.D.; Halilbasic, E.; Trauner, M. Pathophysiologic basis for alternative therapies for cholestasis. Liver Biol. Pathobiol. 2020, 364–377.
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218.
- Kowdley, K.V.; Luketic, V.; Chapman, R.; Hirschfield, G.M.; Poupon, R.; Schramm, C.; Vincent, C.; Rust, C.; Pares, A.; Mason, A.; et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology 2018, 67, 1890–1902.
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N. Engl. J. Med. 2016, 375, 631–643.
- Reig, A.; Sese, P.; Pares, A. Effects of bezafibrate on outcome and pruritus in primary biliary cholangitis with suboptimal ursodeoxycholic acid response. Am. J. Gastroenterol. 2018, 113, 49–55.
- Dejman, A.; Clark, V.; Martin, P.; Levy, C. Fenofibrate improves alkaline phosphatase in primary sclerosing cholangitis. Gastroenterology 2013, 144, S1028–S1029.
- Hommes, D.W.; Erkelens, W.; Ponsioen, C.; Stokkers, P.; Rauws, E.; van der Spek, M.; ten Kate, F.; van Deventer, S.J. A double-blind, placebo-controlled, randomized study of infliximab in primary sclerosing cholangitis. J. Clin. Gastroenterol. 2008, 42, 522–526.
- Hirschfield, G.M.; Gershwin, M.E.; Strauss, R.; Mayo, M.J.; Levy, C.; Zou, B.; Johanns, J.; Nnane, I.P.; Dasgupta, B.; Li, K.; et al. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: A proof-of-concept study. Hepatology 2016, 64, 189–199.
- Santiago, P.; Scheinberg, A.R.; Levy, C. Cholestatic liver diseases: New targets, new therapies. Therap. Adv. Gastroenterol. 2018, 11, 1756284818787400.
- Cai, S.Y.; Li, M.; Boyer, J.L. The role of bile acid-mediated inflammation in cholestatic liver injury. Liver Biol. Pathobiol. 2020, 728–736.
- Bull, L.N.; Thompson, R.J. Progressive familial intrahepatic cholestasis. Clin. Liver Dis. 2018, 22, 657–669.
- Zollner, G.; Trauner, M. Mechanisms of cholestasis. Clin. Liver Dis. 2008, 12, 1–26.
- Carbone, M.; Mells, G.F.; Pells, G.; Dawwas, M.F.; Newton, J.L.; Heneghan, M.A.; Neuberger, J.M.; Day, D.B.; Ducker, S.J.; Consortium, U.P.; et al. Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastroenterology 2013, 144, 560–569.e7.
- Lammers, W.J.; van Buuren, H.R.; Hirschfield, G.M.; Janssen, H.L.; Invernizzi, P.; Mason, A.L.; Ponsioen, C.Y.; Floreani, A.; Corpechot, C.; Mayo, M.J.; et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: An international follow-up study. Gastroenterology 2014, 147, 1338–1349.e5.
- Trivedi, P.J.; Lammers, W.J.; van Buuren, H.R.; Pares, A.; Floreani, A.; Janssen, H.L.; Invernizzi, P.; Battezzati, P.M.; Ponsioen, C.Y.; Corpechot, C.; et al. Stratification of hepatocellular carcinoma risk in primary biliary cirrhosis: A multicentre international study. Gut 2016, 65, 321–329.
- European Association for the Study of the Liver. Electronic address, e.e.e.; European Association for the Study of the, L. Easl clinical practice guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172.
- Gulamhusein, A.F.; Hirschfield, G.M. Primary biliary cholangitis: Pathogenesis and therapeutic opportunities. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 93–110.
- Beuers, U.; Gershwin, M.E.; Gish, R.G.; Invernizzi, P.; Jones, D.E.; Lindor, K.; Ma, X.; Mackay, I.R.; Pares, A.; Tanaka, A.; et al. Changing nomenclature for pbc: From ‘cirrhosis’ to ‘cholangitis’. Gastroenterology 2015, 149, 1627–1629.
- Kaplan, M.M.; Gershwin, M.E. Primary biliary cirrhosis. N. Engl. J. Med. 2005, 353, 1261–1273.
- Dyson, J.K.; Wilkinson, N.; Jopson, L.; Mells, G.; Bathgate, A.; Heneghan, M.A.; Neuberger, J.; Hirschfield, G.M.; Ducker, S.J.; Consortium, U.-P.; et al. The inter-relationship of symptom severity and quality of life in 2055 patients with primary biliary cholangitis. Aliment. Pharmacol. Ther. 2016, 44, 1039–1050.
- Mells, G.F.; Pells, G.; Newton, J.L.; Bathgate, A.J.; Burroughs, A.K.; Heneghan, M.A.; Neuberger, J.M.; Day, D.B.; Ducker, S.J.; Sandford, R.N.; et al. Impact of primary biliary cirrhosis on perceived quality of life: The uk-pbc national study. Hepatology 2013, 58, 273–283.
- Li, T.; Chiang, J.Y. Bile acid metabolism in health and disease: An update. Liver Biol. Pathobiol. 2020, 269–285.
- Gonzalez, R.S.; Washington, K. Primary biliary cholangitis and autoimmune hepatitis. Surg. Pathol. Clin. 2018, 11, 329–349.
- Boonstra, K.; Kunst, A.E.; Stadhouders, P.H.; Tuynman, H.A.; Poen, A.C.; van Nieuwkerk, K.M.; Witteman, E.M.; Hamann, D.; Witteman, B.J.; Beuers, U.; et al. Rising incidence and prevalence of primary biliary cirrhosis: A large population-based study. Liver Int. 2014, 34, e31–e38.
- Dahlqvist, G.; Gaouar, F.; Carrat, F.; Meurisse, S.; Chazouilleres, O.; Poupon, R.; Johanet, C.; Corpechot, C.; French network of Immunology, L. Large-scale characterization study of patients with antimitochondrial antibodies but nonestablished primary biliary cholangitis. Hepatology 2017, 65, 152–163.
- Griffiths, L.; Dyson, J.K.; Jones, D.E. The new epidemiology of primary biliary cirrhosis. Semin. Liver Dis. 2014, 34, 318–328.
- Lu, M.; Zhou, Y.; Haller, I.V.; Romanelli, R.J.; VanWormer, J.J.; Rodriguez, C.V.; Anderson, H.; Boscarino, J.A.; Schmidt, M.A.; Daida, Y.G.; et al. Increasing prevalence of primary biliary cholangitis and reduced mortality with treatment. Clin. Gastroenterol. Hepatol. 2018, 16, 1342–1350.e1.
- Gulamhusein, A.F.; Juran, B.D.; Lazaridis, K.N. Genome-wide association studies in primary biliary cirrhosis. Semin. Liver Dis. 2015, 35, 392–401.
- Trivedi, P.J.; Hirschfield, G.M. The immunogenetics of autoimmune cholestasis. Clin. Liver Dis. 2016, 20, 15–31.
- Corpechot, C.; Chretien, Y.; Chazouilleres, O.; Poupon, R. Demographic, lifestyle, medical and familial factors associated with primary biliary cirrhosis. J. Hepatol. 2010, 53, 162–169.
- Gershwin, M.E.; Selmi, C.; Worman, H.J.; Gold, E.B.; Watnik, M.; Utts, J.; Lindor, K.D.; Kaplan, M.M.; Vierling, J.M.; Group, U.P.E. Risk factors and comorbidities in primary biliary cirrhosis: A controlled interview-based study of 1032 patients. Hepatology 2005, 42, 1194–1202.
- Hamlyn, A.N.; Macklon, A.F.; James, O. Primary biliary cirrhosis: Geographical clustering and symptomatic onset seasonality. Gut 1983, 24, 940–945.
- Invernizzi, P. Human leukocyte antigen in primary biliary cirrhosis: An old story now reviving. Hepatology 2011, 54, 714–723.
- Juran, B.D.; Lazaridis, K.N. Environmental factors in primary biliary cirrhosis. Semin. Liver Dis. 2014, 34, 265–272.
- Probert, P.M.; Leitch, A.C.; Dunn, M.P.; Meyer, S.K.; Palmer, J.M.; Abdelghany, T.M.; Lakey, A.F.; Cooke, M.P.; Talbot, H.; Wills, C.; et al. Identification of a xenobiotic as a potential environmental trigger in primary biliary cholangitis. J. Hepatol. 2018, 69, 1123–1135.
- Wang, J.J.; Yang, G.X.; Zhang, W.C.; Lu, L.; Tsuneyama, K.; Kronenberg, M.; Vela, J.L.; Lopez-Hoyos, M.; He, X.S.; Ridgway, W.M.; et al. Escherichia coli infection induces autoimmune cholangitis and anti-mitochondrial antibodies in non-obese diabetic (nod).B6 (idd10/idd18) mice. Clin. Exp. Immunol. 2014, 175, 192–201.
- Seidel, D.; Eickmeier, I.; Kuhl, A.A.; Hamann, A.; Loddenkemper, C.; Schott, E. Cd8 t cells primed in the gut-associated lymphoid tissue induce immune-mediated cholangitis in mice. Hepatology 2014, 59, 601–611.
- Li, Y.; Tang, R.; Leung, P.S.C.; Gershwin, M.E.; Ma, X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun. Rev. 2017, 16, 885–896.
- Chen, W.; Wei, Y.; Xiong, A.; Li, Y.; Guan, H.; Wang, Q.; Miao, Q.; Bian, Z.; Xiao, X.; Lian, M.; et al. Comprehensive analysis of serum and fecal bile acid profiles and interaction with gut microbiota in primary biliary cholangitis. Clin. Rev. Allergy Immunol. 2020, 58, 25–38.
- Grant, A.J.; Lalor, P.F.; Salmi, M.; Jalkanen, S.; Adams, D.H. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet 2002, 359, 150–157.
- Jimenez-Dalmaroni, M.J.; Gerswhin, M.E.; Adamopoulos, I.E. The critical role of toll-like receptors--from microbial recognition to autoimmunity: A comprehensive review. Autoimmun. Rev. 2016, 15, 1–8.
- Prince, M.; Chetwynd, A.; Newman, W.; Metcalf, J.V.; James, O.F. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: Follow-up for up to 28 years. Gastroenterology 2002, 123, 1044–1051.
- Christensen, E.; Neuberger, J.; Crowe, J.; Altman, D.G.; Popper, H.; Portmann, B.; Doniach, D.; Ranek, L.; Tygstrup, N.; Williams, R. Beneficial effect of azathioprine and prediction of prognosis in primary biliary cirrhosis. Final results of an international trial. Gastroenterology 1985, 89, 1084–1091.
- Jones, D.E.; Metcalf, J.V.; Collier, J.D.; Bassendine, M.F.; James, O.F. Hepatocellular carcinoma in primary biliary cirrhosis and its impact on outcomes. Hepatology 1997, 26, 1138–1142.
- Dyson, J.K.; Beuers, U.; Jones, D.E.J.; Lohse, A.W.; Hudson, M. Primary sclerosing cholangitis. Lancet 2018, 391, 2547–2559.
- Boonstra, K.; Weersma, R.K.; van Erpecum, K.J.; Rauws, E.A.; Spanier, B.W.; Poen, A.C.; van Nieuwkerk, K.M.; Drenth, J.P.; Witteman, B.J.; Tuynman, H.A.; et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013, 58, 2045–2055.
- Schrumpf, E.; Abdelnoor, M.; Fausa, O.; Elgjo, K.; Jenssen, E.; Kolmannskog, F. Risk factors in primary sclerosing cholangitis. J. Hepatol. 1994, 21, 1061–1066.
- Tischendorf, J.J.; Hecker, H.; Kruger, M.; Manns, M.P.; Meier, P.N. Characterization, outcome, and prognosis in 273 patients with primary sclerosing cholangitis: A single center study. Am. J. Gastroenterol. 2007, 102, 107–114.
- Jepsen, P.; Gronbaek, L.; Vilstrup, H. Worldwide incidence of autoimmune liver disease. Dig. Dis. 2015, 33 (Suppl. 2), 2–12.
- Molodecky, N.A.; Kareemi, H.; Parab, R.; Barkema, H.W.; Quan, H.; Myers, R.P.; Kaplan, G.G. Incidence of primary sclerosing cholangitis: A systematic review and meta-analysis. Hepatology 2011, 53, 1590–1599.
- Bambha, K.; Kim, W.R.; Talwalkar, J.; Torgerson, H.; Benson, J.T.; Therneau, T.M.; Loftus, E.V., Jr.; Yawn, B.P.; Dickson, E.R.; Melton, L.J., 3rd. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a united states community. Gastroenterology 2003, 125, 1364–1369.
- Escorsell, A.; Pares, A.; Rodes, J.; Solis-Herruzo, J.A.; Miras, M.; de la Morena, E. Epidemiology of primary sclerosing cholangitis in spain. Spanish association for the study of the liver. J. Hepatol. 1994, 21, 787–791.
- Lindkvist, B.; Benito de Valle, M.; Gullberg, B.; Bjornsson, E. Incidence and prevalence of primary sclerosing cholangitis in a defined adult population in sweden. Hepatology 2010, 52, 571–577.
- Liu, J.Z.; Hov, J.R.; Folseraas, T.; Ellinghaus, E.; Rushbrook, S.M.; Doncheva, N.T.; Andreassen, O.A.; Weersma, R.K.; Weismuller, T.J.; Eksteen, B.; et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat. Genet. 2013, 45, 670–675.
- Ji, S.G.; Juran, B.D.; Mucha, S.; Folseraas, T.; Jostins, L.; Melum, E.; Kumasaka, N.; Atkinson, E.J.; Schlicht, E.M.; Liu, J.Z.; et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat. Genet. 2017, 49, 269–273.
- de Vries, A.B.; Janse, M.; Blokzijl, H.; Weersma, R.K. Distinctive inflammatory bowel disease phenotype in primary sclerosing cholangitis. World J. Gastroenterol. 2015, 21, 1956–1971.
- Katt, J.; Schwinge, D.; Schoknecht, T.; Quaas, A.; Sobottka, I.; Burandt, E.; Becker, C.; Neurath, M.F.; Lohse, A.W.; Herkel, J.; et al. Increased t helper type 17 response to pathogen stimulation in patients with primary sclerosing cholangitis. Hepatology 2013, 58, 1084–1093.
- Sabino, J.; Vieira-Silva, S.; Machiels, K.; Joossens, M.; Falony, G.; Ballet, V.; Ferrante, M.; Van Assche, G.; Van der Merwe, S.; Vermeire, S.; et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from ibd. Gut 2016, 65, 1681–1689.
- Wiesner, R.H.; Grambsch, P.M.; Dickson, E.R.; Ludwig, J.; MacCarty, R.L.; Hunter, E.B.; Fleming, T.R.; Fisher, L.D.; Beaver, S.J.; LaRusso, N.F. Primary sclerosing cholangitis: Natural history, prognostic factors and survival analysis. Hepatology 1989, 10, 430–436.
- Bader, T.R.; Beavers, K.L.; Semelka, R.C. Mr imaging features of primary sclerosing cholangitis: Patterns of cirrhosis in relationship to clinical severity of disease. Radiology 2003, 226, 675–685.
- Bjornsson, E.; Lindqvist-Ottosson, J.; Asztely, M.; Olsson, R. Dominant strictures in patients with primary sclerosing cholangitis. Am. J. Gastroenterol. 2004, 99, 502–508.
- Broome, U.; Lofberg, R.; Veress, B.; Eriksson, L.S. Primary sclerosing cholangitis and ulcerative colitis: Evidence for increased neoplastic potential. Hepatology 1995, 22, 1404–1408.
- Chapman, R.; Fevery, J.; Kalloo, A.; Nagorney, D.M.; Boberg, K.M.; Shneider, B.; Gores, G.J.; American Association for the Study of Liver, D. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010, 51, 660–678.
- Claessen, M.M.; Vleggaar, F.P.; Tytgat, K.M.; Siersema, P.D.; van Buuren, H.R. High lifetime risk of cancer in primary sclerosing cholangitis. J. Hepatol. 2009, 50, 158–164.
- European Association for the Study of the, L. Easl clinical practice guidelines: Management of cholestatic liver diseases. J. Hepatol. 2009, 51, 237–267.
- Linder, S.; Soderlund, C. Endoscopic therapy in primary sclerosing cholangitis: Outcome of treatment and risk of cancer. Hepatogastroenterology 2001, 48, 387–392.
- Bergquist, A.; Ekbom, A.; Olsson, R.; Kornfeldt, D.; Loof, L.; Danielsson, A.; Hultcrantz, R.; Lindgren, S.; Prytz, H.; Sandberg-Gertzen, H.; et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J. Hepatol. 2002, 36, 321–327.
- Boberg, K.M.; Bergquist, A.; Mitchell, S.; Pares, A.; Rosina, F.; Broome, U.; Chapman, R.; Fausa, O.; Egeland, T.; Rocca, G.; et al. Cholangiocarcinoma in primary sclerosing cholangitis: Risk factors and clinical presentation. Scand. J. Gastroenterol. 2002, 37, 1205–1211.
- Fevery, J.; Henckaerts, L.; Van Oirbeek, R.; Vermeire, S.; Rutgeerts, P.; Nevens, F.; Van Steenbergen, W. Malignancies and mortality in 200 patients with primary sclerosering cholangitis: A long-term single-centre study. Liver Int. 2012, 32, 214–222.
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323.
- Sawitza, I.; Kordes, C.; Reister, S.; Haussinger, D. The niche of stellate cells within rat liver. Hepatology 2009, 50, 1617–1624.
- Zhang, Q.D.; Xu, M.Y.; Cai, X.B.; Qu, Y.; Li, Z.H.; Lu, L.G. Myofibroblastic transformation of rat hepatic stellate cells: The role of notch signaling and epithelial-mesenchymal transition regulation. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4130–4138.
- Spee, B.; Carpino, G.; Schotanus, B.A.; Katoonizadeh, A.; Vander Borght, S.; Gaudio, E.; Roskams, T. Characterisation of the liver progenitor cell niche in liver diseases: Potential involvement of wnt and notch signalling. Gut 2010, 59, 247–257.
- Tanriverdi, G.; Kaya-Dagistanli, F.; Ayla, S.; Demirci, S.; Eser, M.; Unal, Z.S.; Cengiz, M.; Oktar, H. Resveratrol can prevent ccl(4)-induced liver injury by inhibiting notch signaling pathway. Histol. Histopathol. 2016, 31, 769–784.
- Chen, Y.; Zheng, S.; Qi, D.; Zheng, S.; Guo, J.; Zhang, S.; Weng, Z. Inhibition of notch signaling by a gamma-secretase inhibitor attenuates hepatic fibrosis in rats. PLoS ONE 2012, 7, e46512.
- Adams, J.M.; Jafar-Nejad, H. The roles of notch signaling in liver development and disease. Biomolecules 2019, 9, 608.
- Zhang, K.; Han, X.; Zhang, Z.; Zheng, L.; Hu, Z.; Yao, Q.; Cui, H.; Shu, G.; Si, M.; Li, C.; et al. The liver-enriched lnc-lfar1 promotes liver fibrosis by activating tgfbeta and notch pathways. Nat. Commun. 2017, 8, 144.
- Aimaiti, Y.; Jin, X.; Wang, W.; Chen, Z.; Li, D. Tgf-beta1 signaling regulates mouse hepatic stellate cell differentiation via the jagged1/notch pathway. Life Sci. 2018, 192, 221–230.
- Wallace, K.; Burt, A.D.; Wright, M.C. Liver fibrosis. Biochem. J. 2008, 411, 1–18.
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462.
- Dropmann, A.; Dooley, S.; Dewidar, B.; Hammad, S.; Dediulia, T.; Werle, J.; Hartwig, V.; Ghafoory, S.; Woelfl, S.; Korhonen, H.; et al. Tgf-beta2 silencing to target biliary-derived liver diseases. Gut 2020, 69, 1677–1690.
- Suzuki, K.; Tanaka, M.; Watanabe, N.; Saito, S.; Nonaka, H.; Miyajima, A. P75 neurotrophin receptor is a marker for precursors of stellate cells and portal fibroblasts in mouse fetal liver. Gastroenterology 2008, 135, 270–281.e3.
- Athwal, V.S.; Pritchett, J.; Llewellyn, J.; Martin, K.; Camacho, E.; Raza, S.M.; Phythian-Adams, A.; Birchall, L.J.; Mullan, A.F.; Su, K.; et al. Sox9 predicts progression toward cirrhosis in patients while its loss protects against liver fibrosis. EMBO Mol. Med. 2017, 9, 1696–1710.
- Duan, J.L.; Ruan, B.; Yan, X.C.; Liang, L.; Song, P.; Yang, Z.Y.; Liu, Y.; Dou, K.F.; Han, H.; Wang, L. Endothelial notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology 2018, 68, 677–690.
- Zhu, C.; Kim, K.; Wang, X.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M.; et al. Hepatocyte notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018, 10, eaat0344.
- Fabris, L.; Spirli, C.; Cadamuro, M.; Fiorotto, R.; Strazzabosco, M. Emerging concepts in biliary repair and fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G102–G116.
- Planas-Paz, L.; Sun, T.; Pikiolek, M.; Cochran, N.R.; Bergling, S.; Orsini, V.; Yang, Z.; Sigoillot, F.; Jetzer, J.; Syed, M.; et al. Yap, but not rspo-lgr4/5, signaling in biliary epithelial cells promotes a ductular reaction in response to liver injury. Cell Stem Cell 2019, 25, 39–53.e10.
- Furth, N.; Aylon, Y.; Oren, M. P53 shades of hippo. Cell Death Differ. 2018, 25, 81–92.
- Bird, T.G.; Lu, W.Y.; Boulter, L.; Gordon-Keylock, S.; Ridgway, R.A.; Williams, M.J.; Taube, J.; Thomas, J.A.; Wojtacha, D.; Gambardella, A.; et al. Bone marrow injection stimulates hepatic ductular reactions in the absence of injury via macrophage-mediated tweak signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 6542–6547.
- Jakubowski, A.; Ambrose, C.; Parr, M.; Lincecum, J.M.; Wang, M.Z.; Zheng, T.S.; Browning, B.; Michaelson, J.S.; Baetscher, M.; Wang, B.; et al. Tweak induces liver progenitor cell proliferation. J. Clin. Investig. 2005, 115, 2330–2340.
- Machado, M.V.; Diehl, A.M. Hedgehog signalling in liver pathophysiology. J. Hepatol. 2018, 68, 550–562.
- Strazzabosco, M.; Fabris, L. Notch signaling in hepatocellular carcinoma: Guilty in association! Gastroenterology 2012, 143, 1430–1434.
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 2008, 118, 3331–3342.
- Xie, G.; Karaca, G.; Swiderska-Syn, M.; Michelotti, G.A.; Kruger, L.; Chen, Y.; Premont, R.T.; Choi, S.S.; Diehl, A.M. Cross-talk between notch and hedgehog regulates hepatic stellate cell fate in mice. Hepatology 2013, 58, 1801–1813.
- Stasiulewicz, M.; Gray, S.D.; Mastromina, I.; Silva, J.C.; Bjorklund, M.; Seymour, P.A.; Booth, D.; Thompson, C.; Green, R.J.; Hall, E.A.; et al. A conserved role for notch signaling in priming the cellular response to shh through ciliary localisation of the key shh transducer smo. Development 2015, 142, 2291–2303.
- He, F.; Guo, F.C.; Li, Z.; Yu, H.C.; Ma, P.F.; Zhao, J.L.; Feng, L.; Li, W.N.; Liu, X.W.; Qin, H.Y.; et al. Myeloid-specific disruption of recombination signal binding protein jkappa ameliorates hepatic fibrosis by attenuating inflammation through cylindromatosis in mice. Hepatology 2015, 61, 303–314.
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53.
- Monga, S.P. Beta-catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology 2015, 148, 1294–1310.
- Ramirez, T.; Li, Y.M.; Yin, S.; Xu, M.J.; Feng, D.; Zhou, Z.; Zang, M.; Mukhopadhyay, P.; Varga, Z.V.; Pacher, P.; et al. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J. Hepatol. 2017, 66, 601–609.
- Roeb, E. Matrix metalloproteinases and liver fibrosis (translational aspects). Matrix Biol. 2018, 68-69, 463–473.
- Beljaars, L.; Daliri, S.; Dijkhuizen, C.; Poelstra, K.; Gosens, R. Wnt-5a regulates tgf-beta-related activities in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G219–G227.
- Carpino, G.; Nobili, V.; Renzi, A.; De Stefanis, C.; Stronati, L.; Franchitto, A.; Alisi, A.; Onori, P.; De Vito, R.; Alpini, G.; et al. Macrophage activation in pediatric nonalcoholic fatty liver disease (nafld) correlates with hepatic progenitor cell response via wnt3a pathway. PLoS ONE 2016, 11, e0157246.
- Ma, Z.G.; Lv, X.D.; Zhan, L.L.; Chen, L.; Zou, Q.Y.; Xiang, J.Q.; Qin, J.L.; Zhang, W.W.; Zeng, Z.J.; Jin, H.; et al. Human urokinase-type plasminogen activator gene-modified bone marrow-derived mesenchymal stem cells attenuate liver fibrosis in rats by down-regulating the wnt signaling pathway. World J. Gastroenterol. 2016, 22, 2092–2103.
- Xiong, W.J.; Hu, L.J.; Jian, Y.C.; Wang, L.J.; Jiang, M.; Li, W.; He, Y. Wnt5a participates in hepatic stellate cell activation observed by gene expression profile and functional assays. World J. Gastroenterol. 2012, 18, 1745–1752.
- Cheng, J.H.; She, H.; Han, Y.P.; Wang, J.; Xiong, S.; Asahina, K.; Tsukamoto, H. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G39–G49.
- Lai, K.K.Y.; Kweon, S.M.; Chi, F.; Hwang, E.; Kabe, Y.; Higashiyama, R.; Qin, L.; Yan, R.; Wu, R.P.; Lai, K.; et al. Stearoyl-coa desaturase promotes liver fibrosis and tumor development in mice via a wnt positive-signaling loop by stabilization of low-density lipoprotein-receptor-related proteins 5 and 6. Gastroenterology 2017, 152, 1477–1491.
- Wang, J.N.; Li, L.; Li, L.Y.; Yan, Q.; Li, J.; Xu, T. Emerging role and therapeutic implication of wnt signaling pathways in liver fibrosis. Gene 2018, 674, 57–69.
- Pradhan-Sundd, T.; Kosar, K.; Saggi, H.; Zhang, R.; Vats, R.; Cornuet, P.; Green, S.; Singh, S.; Zeng, G.; Sundd, P.; et al. Wnt/beta-catenin signaling plays a protective role in the mdr2 knockout murine model of cholestatic liver disease. Hepatology 2020, 71, 1732–1749.
- Minden, A.; Karin, M. Regulation and function of the jnk subgroup of map kinases. Biochim Biophys Acta 1997, 1333, F85–F104.
- Weston, C.R.; Davis, R.J. The jnk signal transduction pathway. Curr. Opin. Cell Biol. 2007, 19, 142–149.
- Zhao, G.; Hatting, M.; Nevzorova, Y.A.; Peng, J.; Hu, W.; Boekschoten, M.V.; Roskams, T.; Muller, M.; Gassler, N.; Liedtke, C.; et al. Jnk1 in murine hepatic stellate cells is a crucial mediator of liver fibrogenesis. Gut 2014, 63, 1159–1172.
- Kluwe, J.; Pradere, J.P.; Gwak, G.Y.; Mencin, A.; De Minicis, S.; Osterreicher, C.H.; Colmenero, J.; Bataller, R.; Schwabe, R.F. Modulation of hepatic fibrosis by c-jun-n-terminal kinase inhibition. Gastroenterology 2010, 138, 347–359.
- Papa, S.; Bubici, C.; Zazzeroni, F.; Franzoso, G. Mechanisms of liver disease: Cross-talk between the nf-kappab and jnk pathways. Biol. Chem. 2009, 390, 965–976.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
955
Revisions:
2 times
(View History)
Update Date:
30 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No