Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chieh-Yu Shen | + 2766 word(s) | 2766 | 2021-06-10 10:11:06 | | | |
| 2 | Lily Guo | Meta information modification | 2766 | 2021-06-30 02:57:34 | | | | |
| 3 | Lily Guo | Meta information modification | 2766 | 2021-06-30 03:00:56 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Shen, C. Non-Coding RNAs and Rheumatoid arthritis. Encyclopedia. Available online: https://encyclopedia.pub/entry/11475 (accessed on 07 February 2026).
Shen C. Non-Coding RNAs and Rheumatoid arthritis. Encyclopedia. Available at: https://encyclopedia.pub/entry/11475. Accessed February 07, 2026.
Shen, Chieh-Yu. "Non-Coding RNAs and Rheumatoid arthritis" Encyclopedia, https://encyclopedia.pub/entry/11475 (accessed February 07, 2026).
Shen, C. (2021, June 29). Non-Coding RNAs and Rheumatoid arthritis. In Encyclopedia. https://encyclopedia.pub/entry/11475
Shen, Chieh-Yu. "Non-Coding RNAs and Rheumatoid arthritis." Encyclopedia. Web. 29 June, 2021.
Copy Citation
Rheumatoid arthritis (RA) is a typical autoimmune-mediated rheumatic disease presenting as a chronic synovitis in the joint.
rheumatoid arthritis
non-coding RNA
1. Introduction
Rheumatoid arthritis (RA) originates from dysregulated immune responses primarily involving the synovial tissues of joints. The abnormal immune responses in the synovial tissues are believed to be triggered by complex interactions between genetic background and environmental factors. Thereby, rheumatoid synovitis is the result of the chronic inflammation elicited by the interactions among innate immune cells including monocytes/macrophages, dendritic cells (DCs), mast cells, natural killer (NK) cells and polymorphonuclear neutrophils (PMNs), and adaptive immune cells including B and T lymphocytes, and fibroblast-like synoviocytes (FLSs). Arbitrarily, the chronic synovial inflammation in RA can be defined as an imbalance between proinflammatory and anti-inflammatory cytokines with preponderance in proinflammatory ones including interleukins (ILs) IL-1β, IL-6, IL-8, IL-17, and tumor necrosis factor (TNF) TNF-α [1]. In addition, the activation, proliferation, and differentiation of B cells to plasma cells lead to the production of a variety of autoantibodies. Among them, the characteristic ones such as rheumatoid factors (RFs) (anti-denatured IgG antibodies) and anti-citrullinated protein antibodies (ACPAs) are highly associated with the pathogenesis and disease activity of RA [2][3]. Well-documented upstream genetic factors implicated in RA pathogenesis include both human histocompatibility antigens (i.e., human leukocyte antigen, HLA, such as HLA-DR4, HLA-DRB1) and non-HLA genes (PTPN22, CTLA-4, TRAF1-C5, STAT4, PADI4) [3]. In addition, a setting of environmental factors such as infections (Epstein–Barr virus), chemicals (pesticides), heavy metals (organic mercury and cadmium), cigarette smoking, and lifestyle factors (socio-economic status and psychological factors) are all established risk elements in triggering RA [4]. However, the most important discovery regarding its pathogenetic factors has been the epigenetic regulation of gene expression which mainly involves non-coding RNAs (ncRNAs). These non-translated small nucleotide sequences can be arbitrarily classified as microRNAs (miRs) with 20–24 nt. in size, and long non-coding RNAs (lncRNAs) with <300 nt. in size. These ncRNAs are not housekeeping molecules but can act as post-transcriptional regulators for mRNA expression by binding to 3′-untranslated regions (3′-UTR) of protein-coding genes [5]. Such binding can be further modulated through a ribonucleoprotein complex called “RNA induced silencing complex (RISC)” [6] where argonaute (Ago) proteins become the major catalytic component. The ncRNAs bound to Ago can guide RISC to the consequent complementary target sites [7]. For further understanding of the immunopathological mechanisms of RA, it is worthwhile looking into the immunopathological roles of the two characteristic autoantibodies, RFs and ACPAs, relevant to the development of RA before going into the detailed discussion of aberrant ncRNA expression in patients with RA.
2. Immunopathologic Roles of RFs and ACPAs in Patients with RA
2.1. Immunopathologic Roles of RFs in RA
Although RFs, one of the most important biomarkers for the diagnosis of RA, are found in 75–85% of patients with RA [8], many other autoimmune diseases including systemic lupus erythematosus (SLE), primary Sjögren’s syndrome (SjS), and mixed connective tissue disease (MCTD) also exhibit RFs. Besides, chronic infections such as hepatitis B virus (HBV), hepatitis C virus (HCV), bacterial endocarditis, leprosy, and tuberculosis (TB) can also stimulate RF production in serum. Soltys et al. [9] revealed that a unique subclass of RFs, anti-agalactosyl IgG (Gal (0)) RF can be generated in the early stage which correlates with severe joint erosion in patients with RA [10][11], similar to ACPAs. Based on these findings, Lu et al. [12] tried to compare anti-Gal (0) RFs, ordinary RFs, and ACPA antibodies in the differential diagnosis of RA and its mimics including SLE, Sjögren’s syndrome, HBV hepatitis, or HCV hepatitis. These authors found that the serum titer of anti-Gal (0) IgG was much higher in RA than in the mimicking diseases. Nevertheless, ACPAs remain the most specific autoantibody for diagnosing RA and evaluating its activity.
2.2. Immunopathologic Roles of ACPAs in RA
Masson-Bessiere et al. [13] first discovered that anti-filaggrin autoantibodies are produced primarily by the local plasma cells in rheumatoid pannus. These autoantibodies against the citrullinated fibrin constituent were found concentrated in the synovial fluid rather than in the plasma [14]. Interestingly, these ACPAs are detectable several years before the overt joint inflammation [15]. The ACPAs are currently recognized as highly specific for RA and can be detected in 75–85% RA sera [16].
It is well recognized that macrophage-derived TNF-α is an upstream proinflammatory cytokine in eliciting inflammation in patients with RA. For testing whether ACPAs are involved in the TNF-α production by macrophages, Clavel et al. [17] developed an in vitro human model in which monocyte-derived macrophages were incubated with ACPA-containing immune complex (ACPA-ICs) generated by mixing RA-derived ACPA and citrullinated fibrin-derived peptides. The authors demonstrated that ACPA-ICs induced a dose-dependent TNF-α secretion from macrophages via engagement of IgG-Fc receptor IIa (FcγRII) on the surface of macrophages. In contrast, Lu et al. [18] clearly demonstrated that RA-derived ACPAs could directly bind to surface-expressed citrullinated glucose-regulated protein 78 (Grp78 with a molecular weight of 72 kDa) on monocytes/macrophages as one of the cognate antigens for ACPAs. The binding activated ERK1/2-JNK-NF-κB signaling pathway and stimulated TNF-α production from monocytes/macrophages [19]. Furthermore, Lai et al. [20] demonstrated that ACPAs obtained from patients with RA could suppress let-7a expression in monocytes and facilitated the inflammatory responses in these patients. In addition, the same group confirmed that ACPAs could react with citrullinated HSP60 expressed on mature human Saos-2 osteoblast cell line to promote IL-6 and IL-8 gene expression as well as apoptosis through Toll-like receptor 4 (TLR4) signaling [21]. These results suggest that ACPAs can directly induce joint inflammation and bone damage in patients with RA.
In addition, investigators demonstrated that ACPAs could promote inflammatory responses through complement activation [21], neutrophil extracellular trap (NET) formation [22][23], and direct binding with osteoclasts [24] and joint chondrocytes [25] to mediate rheumatoid pathogenesis. The pathologic role of ACPAs was critically reviewed by Yu et al. [26] and Wu et al. [27]. Involvement of ACPAs in the pathogenesis of RA is shown in Figure 1.
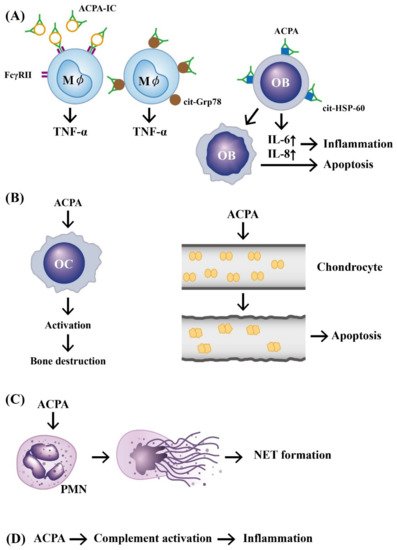
Figure 1. The mechanisms of APCA in eliciting development of rheumatoid arthritis. (A) Preformed immune complexes composed of circulating ACPA and citrullinated proteins bind to FcγR or bind directly to the surface-expressed cit-GRP78 on the macrophage. These two types of binding can stimulate TNF-α production from macrophages. In addition, a direct binding of ACPA to cit-HSP-60 on OB activates IL-6 and IL-8 secretion from OB and subsequently induces cell apoptosis and inflammation; (B) ACPA can directly activate OC to induce bone destruction and elicit neighboring chondrocyte apoptosis after its binding; (C) ACPA can bind to neutrophils and induce NETosis; (D) ACPA can activate the complement system to induce inflammation. ACPA: anti-citrullinated protein antibody (green in color). ACPA: anti-citrullinated protein antibody, OB: osteoblast, OC: osteoclast, Mϕ: macrophage, PMN: polymorphonuclear neutrophil, FcγR: IgG Fragment C receptor, IC: immune complex, TNF: tumor necrosis factor, IL: interleukin, cit-GRP78: citrullinated glucose-regulated protein 78, cit-HSP-60: citrullinated heat shock protein 60, NET: neutrophil extracellular trap.
3. The Effects of Rheumatoid Inflammation-Induced Aberrant Non-Coding RNA Expression on the Modification of Biological Behavior in Fibroblast-Like Synoviocytes (FLSs) of RA Patients (RA-FLSs)
The specific fibroblastic lining cells in the synovial membrane of the joints are called FLSs. These cells play an essential role in the homeostasis of synovial joints by secreting synovial fluid (SF) components for maintaining normal physiological condition [28][29]. However, in pathological conditions such as chronic inflammation, FLSs lose their contact inhibition potential and obtain a high magnitude of proliferation, decreased apoptosis and increased production of cytokines/chemokines, adhesion molecules, and MMPs. These aberrant biological behavioral changes contribute to the invasive cancer-like phenotype behaviors of the FLSs with subsequent pannus formation in rheumatoid joints [30][31]. We will dissect the effect of aberrant epigenetic changes in chronic rheumatoid inflammation on the biological behavioral changes of RA-FLSs and their targeting molecules in the following subsections in detail. In general, the normal biological behaviors of RA-FLSs can be enhanced, inhibited, or regulated by chronic rheumatoid inflammation-induced abnormal ncRNA expressions.
3.1. Derangement of the Biological Behaviors of RA-FLSs by Aberrantly Expressed miRs in the Milieu of Chronic Rheumatoid Inflammation
RA-FLSs are found to share some features with cancer cells including tumor-like migration or invasion, and resistance to apoptosis. These properties render RA a characteristic of spreading and inflammation-induced destruction to other distant joints [32][33]. It is conceivable that synoviocyte proliferation, invasion, and migration are essential for the RA pathology. Chen et al. [34] found that miR-21 was overexpressed in patients with RA and collagen-induced arthritis (CIA) in rat models. They showed that enhanced cell proliferation of FLSs facilitated NF-κB nuclear translocation to transduce the NF-κB signaling pathway. Besides, Huang et al. [35] demonstrated that miR-26a-5p expression was higher than that in osteoarthritis. Overexpression of miR-26a-5p in RA-FLSs promoted cell proliferation, G1/S transition, cell invasion and apoptosis resistance. The 3′-UTR of phosphatase and tensin homolog (PTEN) was directly targeted and the activation of phosphoinositide 3-kinase (PI3K)/AKT pathway was observed. Yu et al. [36] confirmed that hypoxia-induced miR-191 expression was increased in RA-FLS that could increase cellular proliferation via inducing the C/EBPβ signaling pathway.
3.2. Inhibition on Biological Behaviors of RA-FLS by Aberrantly Expressed-miRs in Chronic Rheumatoid Inflammation
Shi et al. [37] found that miR-27a was markedly downregulated in the serum, synovial tissue, and FLSs of RA patients whereas follistatin-like protein 1 (FSTL1) was conversely upregulated. The authors concluded that miR-27a could inhibit cell migration and invasion of RA-FLSs by targeting FSTL1 and restrained the TLR4-NF-κB signaling pathway. Li et al. [38] reported that RA synovial tissues had significant lower levels of miR-19s than controls. The overexpression of miR-19s triggered increase in caspase-3 activity and Bax/Bcl-2 ratio via direct targeting of caveolin 1 (CAV1) in causing cell apoptosis. Wei et al. [39] found that significant lower miR-20a expression and higher STAT3, pSTAT3, and Ki-67 expression in RA synovial tissues enhanced FLS proliferation and reduced apoptosis. Liu et al. [40] observed that miR-29a was markedly downregulated in serum, synovial tissues, and FLS of RA patients. STAT3 was identified to be a direct target of miR-29a in RA-FLS to suppress cell proliferation. Wangyang et al. [41] disclosed downregulated miR-199a-3p expression in RA-FLS. Further studies by the group revealed that the miR-199a-3p could inhibit proliferation and induced apoptosis of RA-FLS via targeting retinoblastoma 1 pathway. Wei et al. [42] demonstrated that poorly expressed miR-101-3p and highly expressed prostaglandin-endoperoxide synthase 2 (PTGS2) in the synovial tissues of RA patients and rat RA models reduced synoviocyte apoptosis and enhanced inflammation. Yang et al. [43] demonstrated that miR-124a suppressed the viability and proliferation of CIA mouse FLSs via the PI3K/AKT/NF-κB signaling pathway. Therefore, miR-124a can inhibit the expression of proinflammatory cytokines, TNF-α and IL-6. Lin et al. [44] confirmed that the miR-320a expression levels were lower in RA synovial tissues than in controls. The miR-320a attenuated proliferation of and promoted apoptosis of RA-FLSs through inhibition of the MAPK-ERK1/2 signaling pathway. Wang et al. [45] discovered that miR-410-3p expression levels were declined in both synovium and FLSs from RA patients. Yingyang 1 (YY1) protein was verified as a direct target of miR-410-3p. Li et al. [46] found that the miR-506 expression levels were significantly lower in the synovial tissues and FLSs in RA patients. The target molecule of miR-506 was proven to be TLR4. Wang et al. [47] discovered that miR-431-5p was downregulated in synovial tissues and FLSs of patients with RA. This miR could directly target the X-lined inhibitor of apoptosis (XIAP) in RA-FLS, suggesting its potential in the treatment of RA. Wang et al. [48] identified a novel miR-141-3p and forkhead box protein C1 (FoxC1)/β-catenin axis that could modulate the inflammation, proliferation, migration, and invasion of RA-FLSs in vivo and in vitro.
3.3. Enhancement on Biological Behaviors of RA-FLS by Aberrantly Expressed-lncRNAs in Chronic Rheumatoid Inflammation
Some lncRNA expressions in cancer cells could promote tumor cell migration and invasion. Similarly, these cancer cell-associated lncRNAs may also be implicated in the pathological behavior of FLSs in patients with RA.
Ye et al. [49] found that lncRNA, ZFAS1, expression was increased in synovial tissues and FLSs from RA patients. ZFAS1 could directly interact with miR-27a by exerting a sponge-like effect to promote migration and invasion of RA-FLSs. Later, Mo et al. [50] found that a newly identified functional lncRNA, GAPLINC, in the oncologic investigation, displayed a high degree of expression in RA-FLSs. The high expression renders RA-FLSs significantly increased in cell proliferation, invasion, migration, and proinflammatory cytokine production. These changes in biological behaviors of RA-FLS are quite similar to the suppression of miR-382 and miR-575 expression by an miR sponging agent. In addition, these authors proved that GAPLINC could promote the tumor-like behaviors of RA-FLS in miR-382-5p- and miR-575-dependent manners. Bi et al. [51] identified that a long intergenic lncRNA162 (LINC00162), also known as lncPISCAR (a p38 inhibited squamous cell carcinoma associated lncRNA), had higher expression levels in RA-FLSs and RA synovial fluid. The lncRNA PISCAR can promote tumor-like behaviors of RA-FLSs through sponging miR-4701-5p. Wang et al. [52] investigated an lncRNA PVT1 (plasmacytoma variant translocation 1) and found this cancer-associated lncRNA was overexpressed in synovial tissue from RA patients. In addition, both PVT1 and SCUBE2 (signal peptide-CUB-EGF-like containing protein 2) were elevated concomitantly whereas miR-534 was reduced in synovial tissues from RA patients or CIA mice. It was also confirmed that lncRNA PVT1 specifically binds to miR-543, and then negatively regulates SCUBE2 expression. Thereby, lncRNA PVT1 can promote RA-FLS proliferation and its IL1-β secretion, while inhibiting its apoptosis. Alternatively, Tang et al. [53] demonstrated that PVT1 was significantly increased whereas miR-145-5p was decreased in RA synovial tissues. Interestingly, miR-145-5p has been found to be a target miR of PVT1 as well which can be induced by TNF-α while it can be suppressed concomitantly on its own by TNF-α. As an oncogene in many human cancers, lncRNA NEAT1 is upregulated in RA synovial tissues and FLSs as reported by Wang et al. [54]. These authors disclosed that the upregulation of lncRNA NEAT1 can promote cell proliferation by inducing transition of cell cycle from the S phase to the G2/M phase, and suppresses apoptosis of RA-FLSs. Further studies revealed that NEAT1 could directly bind to miR-410-3p (sponging effect) and negatively modulate its expression, but positively regulate YingYang1 (YY1, a miR-410-3p target protein). Xiao et al. [55] also confirmed that lncRNA NEAT1 is upregulated and miR-204 is downregulated in RA synovial tissues and TNF-α-treated RA-FLSs. The addition of TNF-α enhanced lncNEAT1 levels but decreases miR-204-5p expression in cultured RA-FLSs. The knockdown of NEAT1 attenuated TNF-α-induced RA-FLS cell proliferation and proinflammatory cytokine production, while promoting its apoptosis by targeting miR-204-5p via the NF-κB pathway.
The aberrant expression of miRs and lncRNAs in enhancing biological behaviors of RA-FLSs are depicted in Figure 2.
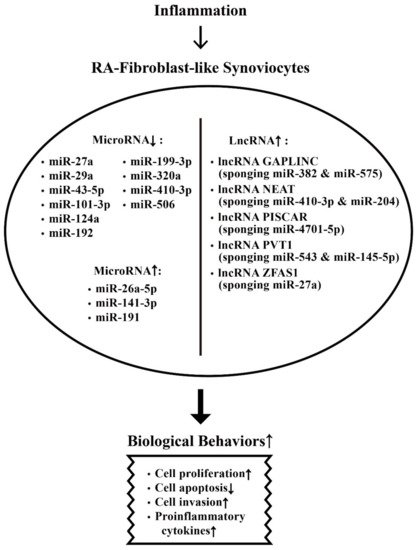
Figure 2. The effects of inflammation on ncRNA expressions and on the biological behavioral changes of rheumatoid arthritis fibroblast-like synoviocytes (RA-FLSs) that are relevant to RA pathogenesis. The biological behavioral changes of RA-FLSs include increased cell proliferation, decreased cell apoptosis, enhanced cell invasion, and augmenting proinflammatory cytokine productions. ↑: Increase, ↓: Decrease.
4. Suppression on Biological Behaviors of RA-FLS by Exosome-Derived ncRNAs from Bone Marrow Derived Mesenchymal Stem Cells (BM-MSCs)
Recently, Cosenza et al. [56] found that both MSCs-released microparticles and exosomes could suppress T- and B-cell-mediated inflammation. The MSC-released exosomes are more potent in protection of RA-FLS from inflammation. Furthermore, Chen et al. [57] reported that MSC-derived exosomes could effectively alleviate the inflammation in experimental RA models partially via the effects of miR-150-5p. Similarly, Zheng et al. [58] found that the MSCs-derived exosomal miR-192-5p could delay inflammatory responses in RA. Meng et al. [59] disclosed that MSC-derived exosomal miR-320a significantly suppressed CIA in vivo by targeting CXC9 expression in RA-FLSs. Su et al. [60] showed that lncRNA HAND2-AS1 (heart and neural crest-derivative expressed 2-antisense RNA1) was expressed at low magnitude in RA synovial tissues. The HAND2-AS1 re-expression could suppress the proliferation, motility, and inflammation of RA-FLSs and trigger their apoptosis through the NF-κB signaling pathway. The same authors further disclosed that MSC-derived exosomal lncRNA HAND2-AS1 suppressed RA-FLS activation via the miR-143-3p/TNFAIP3/NF-κB signaling pathway. These results may provide a novel insight into the potential therapeutic strategy of using MSC-derived exosomal ncRNAs for RA treatment in future.
The effects of MSC-derived exosomal ncRNA on suppressing biological behaviors of RA-FLS and RA inflammation are illustrated in Figure 3.
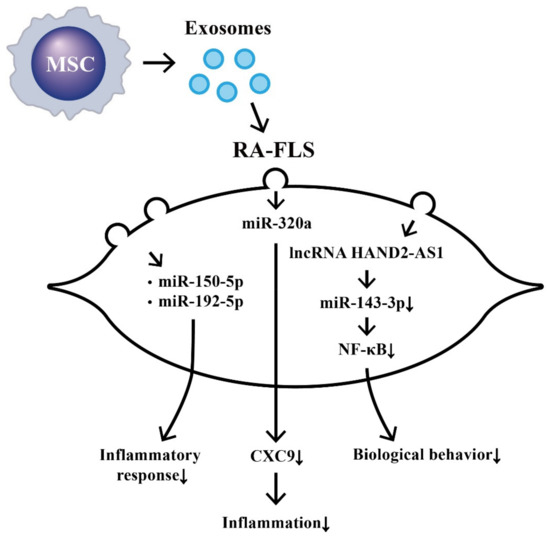
Figure 3. The effects of exosomes released from bone-marrow-derived mesenchymal stem cells (BM-MSCs) on the changes in biological behavior of rheumatoid arthritis fibroblast-like synoviocytes (RA-FLSs). The extruded exosomes containing ncRNAs are enclosed by a lipid bilayer and can move, attach, and merge into the remote RA-FLSs to influence their biological behaviors. Some of the miRs can then suppress inflammatory responses, chemotaxis, and biological behaviors of RA-FLSs.
References
- Mateen, S.; Zafar, A.; Moin, S.; Khan, A.Q.; Zubair, S. Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. Clin. Chim. Acta 2016, 455, 161–171.
- Angelotti, F.; Parma, A.; Cafaro, G.; Capecchi, R.; Alunno, A.; Puxeddu, I. One year in review 2017: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2017, 35, 368–378.
- Zamanpoor, M. The genetic pathogenesis, diagnosis and therapeutic insight of rheumatoid arthritis. Clin. Genet. 2019, 95, 547–557.
- Gourley, M.; Miller, F.W. Mechanisms of disease: Environmental factors in the pathogenesis of rheumatic disease. Nat. Clin. Pract. Rheumatol. 2007, 3, 172–180.
- Zhang, R.; Su, B. Small but influential: The role of microRNAs on gene regulatory network and 3′UTR evolution. J. Genet. Genomics 2009, 36, 1–6.
- Gregory, R.I.; Chendrimada, T.P.; Cooch, N.; Shiekhattar, R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 2005, 123, 631–640.
- Iwasaki, S.; Tomari, Y. Argonaute-mediated translational repression (and activation). Fly 2009, 3, 204–206.
- Ateş, A.; Kinikli, G.; Turgay, M.; Akay, G.; Tokgöz, G. Effects of rheumatoid factor isotypes on disease activity and severity in patients with rheumatoid arthritis: A comparative study. Clin. Rheumatol. 2007, 26, 538–545.
- Soltys, A.J.; Hay, F.C.; Bond, A.; Axford, J.S.; Jones, M.G.; Randen, I.; Thompson, K.M.; Natvig, J.B. The binding of synovial tissue-derived human monoclonal immunoglobulin M rheumatoid factor to immunoglobulin G preparations of differing galactose content. Scand. J. Immunol. 1994, 40, 135–143.
- Young, A.; Sumar, N.; Bodman, K.; Goyal, S.; Sinclair, H.; Roitt, I.; Isenberg, D. Agalactosyl IgG: An aid to differential diagnosis in early synovitis. Arthritis Rheum. 1991, 34, 1425–1429.
- Van Zeben, D.; Rook, G.A.; Hazes, J.M.; Zwinderman, A.H.; Zhang, Y.; Ghelani, S.; Rademacher, T.W.; Breedveld, F.C. Early agalactosylation of IgG is associated with a more progressive disease course in patients with rheumatoid arthritis: Results of a follow-up study. Br. J. Rheumatol. 1994, 33, 36–43.
- Lu, M.-C.; Hsieh, S.-C.; Lai, N.-S.; Li, K.-J.; Wu, C.-H.; Yu, C.-L. Comparison of anti-agalactosyl IgG antibodies, rheumatoid factors, and anti-cyclic citrullinated peptide antibodies in the differential diagnosis of rheumatoid arthritis and its mimics. Clin. Exp. Rheumatol. 2007, 25, 716–721.
- Masson-Bessière, C.; Sebbag, M.; Durieux, J.J.; Nogueira, L.; Vincent, C.; Girbal-Neuhauser, E.; Durroux, R.; Cantagrel, A.; Serre, G. In the rheumatoid pannus, anti-filaggrin autoantibodies are produced by local plasma cells and constitute a higher proportion of IgG than in synovial fluid and serum. Clin. Exp. Immunol. 2000, 119, 544–552.
- Masson- Bessière, C.; Sebbag, M.; Girbal-Neuhauser, E.; Nagueira, L.; Vincent, C.; Senshu, T.; Serre, G. The major synovial targets of the rheumatoid arthritis-specific anti-filaggrin autoantibodies are deiminated forms of the α- and β-chains of fibrin. J. Immunol. 2001, 166, 4177–4184.
- Nielen, M.M.; van Schaardenburg, D.; Reesink, H.W.; van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.M.T.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A.C. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. 2004, 50, 380–386.
- Vincent, C.; Nogueira, L.; Clavel, C.; Sebbag, M.; Serre, G. Autoantibodies to citrullinated proteins: APCA. Autoimmunity 2005, 38, 17–24.
- Clavel, C.; Nogueira, L.; Laurent, L.; Iobagiu, C.; Vincent, C.; Sebbag, M.; Serre, G. Induction of macrophage secretion of tumor necrosis factor α through Fcγ receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum. 2008, 58, 678–688.
- Lu, M.-C.; Lai, N.-S.; Yu, H.-C.; Huang, H.-B.; Hsieh, S.-C.; Yu, C.-L. Anti-citrullinated protein antibodies bind surface-expressed citrullinated Grp78 on monocytes/macrophages and stimulate tumor necrosis factor α production. Arthritis Rheum. 2010, 62, 1213–1223.
- Lu, M.C.; Lai, N.S.; Yin, W.Y.; Yu, H.C.; Huang, H.B.; Tung, C.H.; Huang, K.Y.; Yu, C.L. Anti-citrullinated protein antibodies activated ERK1/2 and JNK mitogen-activated protein kinases via surface-expressed citrullinated Grp78 on mononuclear cells. J. Clin. Immunol. 2013, 33, 558–566.
- Lai, N.S.; Yu, H.C.; Yu, C.L.; Koo, M.; Huang, H.B.; Lu, M.C. Anti-citrullinated protein antibodies suppress let-7a expression in monocytes from patients with rheumatoid arthritis and facilitate the inflammatory responses in rheumatoid arthritis. Immunobiology 2015, 220, 1351–1358.
- Lu, M.H.; Yu, C.L.; Yu, H.C.; Huang, H.B.; Koo, M.; Lai, N.S. Anti-citrullinated protein antibodies promote apoptosis of mature human Saos-2 osteoblasts via cell-surface binding to citrullinated heat shock protein 60. Immunobiology 2016, 221, 76–83.
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra40.
- Pratesi, F.; Dioni, I.; Tommasi, C.; Alcaro, M.C.; Paolini, I.; Barbetti, F.; Boscaro, F.; Panza, F.; Puxeddu, I.; Rovero, P.; et al. Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann. Rheum. Dis. 2014, 73, 1414–1422.
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.-J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802.
- Ge, C.; Tong, D.; Liang, B.; Lönnoblom, E.; Schneider, N.; Hagert, C.; Viljanen, J.; Ayoglu, B.; Stawikowska, R.; Nilsson, P.; et al. Anti-citrullinated protein antibodies cause arthritis by cross-reactivity to joint cartilage. JCI Insight 2017, 2, e93688.
- Yu, H.-C.; Lu, M.-C. The roles of anti-citrullinated protein antibodies in the immunopathogenesis of rheumatoid arthritis. Tzu Chi Med. J. 2019, 31, 5–10.
- Wu, C.Y.; Yang, H.Y.; Lai, J.H. Anti-citrullinated protein antibodies in patients with rheumatoid arthritis: Biological effects and mechanisms of immunopathogenesis. Int. J. Mol. Sci. 2020, 21, 4015.
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255.
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 23–33.
- Ganesan, R.; Rasool, M. Fibroblast-like synoviocytes-dependent effector molecules as a critical mediator for rheumatoid arthritis: Current status and future directions. Int. Rev. Immunol. 2017, 36, 20–30.
- Mousavi, M.J.; Karami, J.; Aslani, S.; Tahmasebi, M.N.; Vaziri, A.S.; Jamshidi, A.; Farhadi, E.; Mahmoudi, M. Transformation of fibroblast-like synoviocytes in rheumatoid arthritis; from friend to foe. Auto Immun. Highlights 2021, 12, 3.
- Ospelt, C.; Neidhart, M.; Gay, R.E.; Gay, S. Synovial activation in rheumatoid arthritis. Front. Biosci. 2004, 9, 2323–2334.
- Zeng, S.; Wang, K.; Huang, M.; Qiu, Q.; Xiao, Y.; Shi, M. Halofuginone inhibits TNF-α-induced the migration and proliferation of fibroblast-like synoviocyte from rheumatoid arthritis patients. Int. Immunopharmacol. 2017, 43, 187–194.
- Chen, Y.; Xian, P.-F.; Yang, L.; Wang, S.-X. MicroRNA-21 promotes proliferation of fibroblast-like synoviocytes through mediation of NF-κB nuclear translocation in a rat-model of collagen-induced rheumatoid arthritis. Biomed. Res. Int. 2016, 9279078.
- Huang, Z.; Xing, S.; Liu, M.; Deng, W.; Wang, Y.; Huang, Z.; Huang, Y.; Huang, X.; Wu, C.; Guo, X.; et al. MiR-26a-5p enhances cells proliferation, invasion, and apoptosis resistance of fibroblast-like synoviocytes in rheumatoid arthritis by regulating PTEN/PI3K/AKT pathway. Biosci. Rep. 2019, 39, BSR20182192.
- Yu, S.; Lu, Y.; Zong, M.; Tan, Q.; Fan, L. Hypoxia-induced miR-91-C/EBPβ signaling regulates cell proliferation and apoptosis of fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Res. Ther. 2019, 21, 78.
- Shi, D.-L.; Shi, G.-R.; Xie, J.; Du, X.-Z.; Yang, H. MicroRNA-27a inhibits cell migration and invasion of fibroblast-like synoviocytes by targeting follistatin-like protein 1 in rheumatoid arthritis. Mol. Cells 2016, 39, 611–618.
- Li, S.; Jin, Z.; Lu, X. MicroRNA-192 suppresses cell proliferation and induces apoptosis in human rheumatoid arthritis fibroblast-like synoviocytes by downregulating caveolin 1. Mol. Cell. Biochem. 2017, 432, 123–130.
- Wei, X.-J.; Li, X.-W.; Lu, J.-L.; Long, Z.-X.; Liang, J.-Q.; Wei, S.-B.; Lu, C.-X.; Lu, W.-Z. MiR-20a regulates fibroblast-like synoviocyte proliferation and apoptosis in rheumatoid arthritis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3886–3893.
- Liu, J.; Fei, D.; Xing, J.; Du, J. MicroRNA-29a inhibits proliferation and induces apoptosis in rheumatoid arthritis fibroblast-like synoviocytes by repressing STAT3. Biomed. Pharmacother. 2017, 96, 173–181.
- Wangyang, Y.; Yi, L.; Wang, T.; Feng, Y.; Liu, G.; Li, D.; Zheng, X. MiR-199a-3p inhibits proliferation and induces apoptosis in rheumatoid arthritis. fibroblast-like synoviocyte via suppressing retinoblastoma 1. Biosci. Rep. 2018, 38, BSR20180982.
- Wei, Q.; Lv, F.; Zhang, H.; Wang, X.; Geng, Q.; Zhang, X.; Li, T.; Wang, S.; Wang, Y.; Cui, Y. MicroRNA-101-3p inhibits fibroblast-like synoviocyte proliferation and inflammation in rheumatoid arthritis by targeting PTGS2. Biosci. Rep. 2020, 40, BSR20191136.
- Yang, B.; Ge, Y.; Zhou, Y.; Wang, J.; Xie, X.; Li, S.; Tang, M.; Xu, L.C.; Tian, J. miR-124a inhibits the proliferation and inflammation in rheumatoid arthritis fibroblast-like synoviocytes via targeting PIK3/NF-κB pathway. Cell. Biochem. Funct. 2019, 37, 208–215.
- Lin, K.; Su, H.-Y.; Jiang, L.-F.; Chu, T.-G.; Li, Z.; Chen, X.-L.; Gao, W.-Y. Influences of miR-320a on proliferation and apoptosis of fibroblast-like synoviocyte in rheumatoid arthritis through targeting MAPK-ERK1/2. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1907–1914.
- Wang, Y.; Jiao, T.; Fu, W.; Zhao, S.; Yang, L.; Xu, N.; Zhang, N. miR-410-3p regulates proliferation and apoptosis of fibroblast-like synovioocytes by targeting YY1 in rheumatoid arthritis. Biomed. Pharmacother. 2019, 119, 109426.
- Li, D.; Zhou, Q.; Hu, G.; Wang, G. MiRNA-506 inhibits rheumatoid arthritis fibroblast-like synoviocytes proliferation and induces apoptosis by targetting TLR4. Biosci. Rep. 2019, 39, BSR20182500.
- Wang, Y.; Zhang, K.; Yuan, X.; Xu, N.; Zhao, S.; Hou, L.; Yang, L.; Zhang, N. miR-431-5p regulates cell proliferation and apoptosis in fibroblast-like synoviocytes in rheumatoid arthritis by targeting XIAP. Arthritis Res. Ther. 2020, 22, 231.
- Wang, J.; Wang, Y.; Zhang, H.; Chang, J.; Lu, M.; Gao, W.; Liu, W.; Li, Y.; Yin, L.; Wang, X.; et al. Identification of a novel microRNA-141-3p/forkhead box C1/β-catenin axis associated with rheumatoid arthritis synovial fibroblast function in vivo and in vitro. Theranostics 2020, 10, 5412–5434.
- Ye, Y.; Gao, X.; Yang, N. LncRNA ZFAS1 promotes cell migration and invasion of fibroblast-like synoviocytes by suppression of miR-27a in rheumatoid arthritis. Hum. Cell 2018, 31, 14–21.
- Mo, B.Y.; Guo, X.H.; Yang, M.R.; Liu, F.; Bi, X.; Liu, Y.; Fang, L.K.; Luo, X.Q.; Wang, J.; Bellanti, J.A.; et al. Long non-coding RNA GAPLINC promotes tumor-like biologic behaviors of fibroblast-like synoviocytes as microRNA sponging in rheumatoid arthritis patients. Front. Immunol. 2018, 9, 702.
- Bi, X.; Guo, X.H.; Mo, B.Y.; Wang, M.L.; Luo, X.Q.; Chen, Y.X.; Liu, F.; Olsen, N.; Pan, Y.F.; Zheng, S.G. LncRNA PICSAR promotes cell proliferation, migration, and invasion of fibroblast-like synoviocytes by sponging miR-4701-5p in rheumatoid arthritis. EBioMedicine 2019, 50, 408–420.
- Wang, J.; Kong, X.; Hu, H.; Shi, S. Knockdown of long non-coding RNA PVT1 induces apoptosis of fibroblast-like synoviocytes through modulating miR-543-dependent SCUBE2 in rheumatoid arthritis. J. Orthop. Surg. Res. 2020, 15, 142.
- Tang, J.; Yi, S.; Liu, Y. Long non-coding RNA PVT1 can regulate the proliferation and inflammatory responses of rheumatoid arthritis fibroblast-like synoviocytes by targeting microRNA-145-5p. Hum. Cell 2020, 33, 1081–1090.
- Wang, Y.; Hou, L.; Yuan, X.; Xu, N.; Zhao, S.; Yang, L.; Zhang, N. LncRNA NEAT1 targets fibroblast-like synoviocyte in rheumatoid arthritis via the miR-410-3p/yy1 axis. Front. Immunol. 2020, 11, 1975.
- Xiao, J.; Wang, R.; Zhou, W.; Cai, X.; Ye, Z. LncRNA NEAT1 regulates the proliferation and production of the inflammatory cytokines in rheumatoid arthritis fibroblast-like synoviocytes by targeting miR-204-5p. Hum. Cell 2021, 34, 372–382.
- Cosenza, S.; Toupet, K.; Maumus, M.; Luz-Crawford, P.; Blanc-Brude, O.; Jorgensen, C.; Noël, D. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics 2018, 8, 1399–1410.
- Chen, Z.; Wang, H.; Xia, Y.; Yang, F.; Lu, Y. Therapeutic potential of mesenchymal cell-derive miRNA-150-5p-expressing exosomes in rheumatoid arthritis mediated by the modulation of MMP14 and VEGF. J. Immunol. 2018, 201, 2472–2482.
- Zheng, J.; Zhu, L.; In, I.I.; Chen, Y.; Jia, N.; Zhu, W. Bone marrow-derived mesenchymal stem cells-secreted exosomal microRNA-192-5p delays inflammatory response in rheumatoid arthritis. Int. Immunopharmacol. 2020, 78, 105985.
- Meng, Q.; Qiu, B. Exosomal microRNA-320a derived from mesenchymal stem cells regulates rheumatoid arthritis fibroblast-like synoviocyte activation by suppressing CXCL9 expression. Front. Physiol. 2020, 11, 441.
- Su, Y.; Liu, Y.; Ma, C.; Guan, C.; Ma, X.; Meng, S. Mesenchymal stem cell-originated exosomal lncRNA HAND2-AS1 impairs rheumatoid arthritis fibroblast-like synoviocyte activation through miR-143-3p/TNFAIP3/NF-κB pathway. J. Orthop. Surg. Res. 2021, 16, 116.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
742
Revisions:
3 times
(View History)
Update Date:
30 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No