In recent years, the role of B cells in neurological disorders has substantially expanded our perspectives on mechanisms of neuroinflammation. The success of B cell-depleting therapies in patients with CNS diseases such as neuromyelitis optica and multiple sclerosis has highlighted the importance of neuroimmune crosstalk in inflammatory processes. While B cells are essential for the adaptive immune system and antibody production, they are also major contributors of pro- and anti-inflammatory cytokine responses in a number of inflammatory diseases. B cells can contribute to neurological diseases through peripheral immune mechanisms, including production of cytokines and antibodies, or through CNS mechanisms following compartmentalization. Emerging evidence suggests that aberrant pro- or anti-inflammatory B cell populations contribute to neurological processes, including glial activation, which has been implicated in the pathogenesis of several neurodegenerative diseases.
1. Introduction
The central nervous system (CNS) was traditionally considered to be a site of strict immune privilege, since there was a perceived lack of lymphatic vessels and professional antigen-presenting cells in the brain parenchyma as well as physical barriers preventing circulating immune cells from entering the CNS. Subsequent studies redefined the concept of immune privilege within the CNS as knowledge of the peripheral immune system and CNS interactions expanded. Although the CNS continues to be considered immune privileged, it is now clear that low numbers of lymphocytes enter CNS lymphatic vessels under healthy steady-state conditions and support CNS immune surveillance
[1]. One conduit of cellular dispersal through the CNS is via the cerebrospinal fluid (CSF). The choroid plexus is the main source of CSF and consists of epithelial cells that form a tight barrier separating the blood and CNS
[2][3]. CSF-interstitial fluid (ISF) drainage follows specific pathways in the CNS, involving the ventricles, subarachnoid space, parenchyma, and subcortical regions, and drain into deep cervical lymph nodes
[4][5]. This functional connection between the CNS and cervical lymph nodes has further supported the concept of neuroimmune crosstalk
[6][7][8][9]. In an injured or inflamed CNS, lymphocytes, including T and B cells, increase by severalfold and can be found throughout the parenchyma, CSF-ISF, and perivascular and meningeal spaces due to antigens draining to lymph nodes or disruptions to the blood-brain barrier
[3][8][10]. The emerging role of the adaptive immune system in the pathophysiology of neurological disorders has led to the successful development of treatments that specifically target the adaptive immune system, including B cell-depleting monoclonal antibodies.
2. B Cells in Neurological Diseases
2.1. Multiple Sclerosis
MS is a chronic inflammatory demyelinating disease characterized by demyelinating lesions in the brain and spinal cord. Although MS is primarily recognized as a white matter disease, both MRI and neuropathological studies of the brain have shown extensive gray matter involvement, particularly of the cortical regions, even at early stages of the disease, and the degree of gray matter involvement is correlated with physical and cognitive symptoms and becomes more fulminant in progressive forms of the disease
[11][12]. In addition to demyelination, microglial and astrocyte activation as well as axonal loss are frequently associated with disease activity. While the disease onset and clinical course may differ, approximately 90% of patients present with relapsing-remitting MS (RRMS) in which patients have periods of relapse (new or worsening neurological symptoms) and remission
[13][14]. After a variable number of years, for most RRMS patients, symptoms eventually worsen with no periods of remission, and this is defined as secondary progressive MS (SPMS)
[13][15][16]. Individuals with accumulating disability from the onset without relapses have a variant of the disease referred to as primary progressive MS (PPMS)
[17][13][18].
Although CNS demyelination is a hallmark of MS, genetic studies showing an association with the MHC class II
HLA-DRB1*15:01 allele
[19][20][21][22] suggest that MS has a critical adaptive immune system component that drives disease. Important advances in our understanding of the pathology of MS have come from the development of animal models that mimic aspects of the human disease. With the realization that MS was predominantly a disease of white matter, initial studies involved the injection of white matter preparations to provoke an immune response and subsequent pathology. While successful, the approach was inconsistent and later refinements including the use of more defined antigens combined with stimulation of the immune system with pertussis toxin resulted in the establishment of experimental allergic encephalomyelitis (EAE). While there are currently a number of variations of EAE, one of the most common models involves injection of peptides of myelin oligodendrocyte glycoprotein (MOG—a major component of CNS myelin) coupled with an adjuvant into an appropriate strain of mice
[23][24][25]. This results in demyelination, predominantly in the spinal cord. The demyelination is associated with immune cell infiltration and glial activation, reminiscent of lesions in MS. Historically, MS has been considered a T cell-mediated disease in large part due to histological observations showing an abundance of T cells in demyelinating lesions
[1]. Furthermore, CD4 and CD8 T cells have been shown to be present in the healthy adult brain parenchyma, where they may be retained after local infection
[26]. A role for T cells in the generation of EAE pathology was further demonstrated through the adoptive transfer of myelin-specific T cells that were able to induce EAE
[27]. These studies guided the field to the development of many of the therapies that are currently available, in particular interferon ß
[28][29]. A potential challenge with the murine EAE models is the influence of species specificity, and to begin addressing these concerns a non-human primate model was developed in the marmoset. In a series of studies, it was shown that selective depletion of B cells resulted in significant reductions in the extent of demyelination in the marmoset EAE model
[30][31]. Indeed, without B cells, white and gray matter damage was significantly reduced, suggesting a necessary role of B cells in disease progression.
The best-known function of B cells is the production of antibodies, and traditionally, it was believed that the role of B cells in MS was the production of autoantibodies directed against myelin proteins. One of the earliest implications of a role for antibodies in MS was the presence of unique immunoglobulins (IgG) in the CSF of approximately 90% of MS patients. Transcriptomic studies later confirmed the source of these IgG’s as clonal B cell populations
[32]. Though target antigens of these clonal IgG fractions have been extensively studied and candidates have been proposed, their function remains uncertain. The presence of these intrathecal IgG fractions, referred to as oligoclonal bands in antibody assays, was however, found not to be specific to MS but also present in neurological conditions, such as encephalitis and neuroborreliosis
[33][34]. Unlike other conditions, such as measles, Herpes simplex virus, and Cytomegalovirus, where the antibodies are directed against epitopes from the causative agents, the significance of the antigens in MS are unknown
[35][36], suggesting either that a MS-specific autoantibody has yet to be identified or more likely rather, the role of B cells in neuroinflammation is not primarily due to antibody production.
The hypothesis that there is an antibody-independent role for B cells in MS is supported by recent successful clinical trials using various anti-CD20 treatments, which target B cells that do not produce antibodies
[17][37][38][39]. The CD20 protein is not expressed on pro-B cells or antibody-secreting plasma cells, however, a clinical trial with rituximab, a monoclonal antibody that selectively targets CD20+ B cells, reported a significant reduction in relapses and gadolinium-enhancing lesions in patients receiving rituximab compared to the placebo group
[37]. Even in primary progressive MS cases, treatment with ocrelizumab, another anti-CD20 antibody, resulted in decreased disability progression and brain lesion volume
[17]. Since disease progression is altered by the depletion of B cells without affecting plasma cells, the most likely interpretation is that B cells play a role in MS pathogenesis through an antibody-independent mechanism (
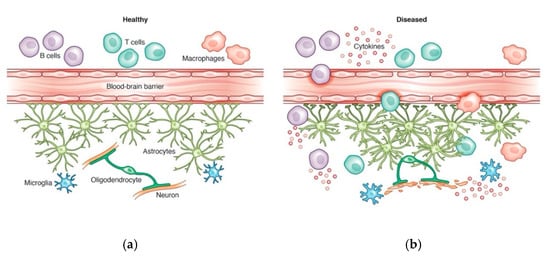
Figure 1).
Figure 1. (a) In the healthy CNS, the blood-brain barrier prevents widespread infiltration of peripheral immune cells, including B cells, T cells, and macrophages. (b) In the diseased, inflamed brain, pro-inflammatory peripheral immune cells may enter the CNS through a dysfunctional blood-brain barrier, allowing cytokine release in the CNS. This increases astrocyte and microglia reactivity, which further propagates the inflammatory cascade, damaging myelin and neurons. Credit: Katie Vicari.
2.2. Parkinson’s Disease
Parkinson’s disease (PD) is a neurodegenerative disorder associated with loss of dopaminergic neurons of the substantia nigra pars compacta in the midbrain, resulting in dopamine deficiency in the nigrostriatal pathway
[40][41][42]. Symptoms include resting tremor, bradykinesia, and impaired posture and balance. Although the exact etiology of PD is unknown, multiple studies have shown an interplay between the CNS and immune system in PD that may result in a dysregulated immune response in the brain, similar to that seen in MS
[43][44][45].
For the past few decades, it has been understood that the pathogenesis in PD is associated with inflammatory responses in the brain. Analysis of
post-mortem brain tissues from patients with PD demonstrated the presence of perivascular deposits of IgG as well as fibrinogen in the striatum, suggesting disruption to the BBB
[46][47][48][49][50]. Analysis of patient samples as well as tissue from mouse models of PD have identified CD8+ and CD4+ T cell infiltration into the CNS but little evidence of B cells in the CNS parenchyma
[51]. Although there is little evidence of widespread CD19+ or CD20+ B cells presence in the CNS of patients with PD, increased levels of antibodies against CNS proteins have been reported
[52][53], suggesting a role for B lineage cells in disease progression. For example, B cell activation can lead to antibody secretion in an inflammatory setting, involving pattern recognition receptor or cytokines released by immune cells. An alternative antibody mediated pathogenic pathway may be the ability of antibodies to initiate or propagate inflammatory reactions by activating the complement system that may result in death of the target cells. Furthermore, activation of the complement system can also lead to microglial activation that can contribute to neuronal loss. Indeed, specific antibodies against dopaminergic neurons have been found in patients with PD
[43][51][52], and these antibodies have been found to colocalize with dopaminergic neurons that are closely associated with FcγR+ microglia
[45], suggesting microglial activation, antibody-mediated phagocytosis of neurons, and release of pro-inflammatory mediators. The role of antibodies in PD is further supported by pre-clinical animal models of PD showing perivascular inflammation and microgliosis after injection of IgG into the substantia nigra of rats
[53]. The same procedure in FcγR(-/-) mice resulted in no microglia activity and nigral injury, suggesting a role for IgG binding in effector cell responses in the CNS. Microglia both respond to as well as propagate inflammation, so it is unclear whether the neuroinflammation was a direct result of IgG presence and whether there is a direct correlation of IgG cytotoxicity and PD disease activity.
Parkinson’s disease is associated with changes in the cellular composition of the immune system
[44][45][54]. For example, analysis of circulating leukocyte populations, including T, B, and natural killer cells, has shown a small reduction in CD4+ T cells as well as B cells in samples from PD patients
[54]. In fact, a continued loss of the T and B cell populations has been reported with clinical stage of disease that was not due to apoptosis
[54], suggesting that disease progression may be influenced by alterations to these populations. The means by which PD influences the cell composition of the immune system is unclear but may not be specific to the disease. In other autoimmune diseases, reduced circulating B cell populations were found to be associated with acute inflammation, possibly regulated by pro-inflammatory cytokines
[55][56]. Further studies are needed to determine the significance of these findings. Although a PD-specific antigen has not yet been identified, it remains possible that B cells play a role in upregulating or downregulating adaptive immune responses by their secretion of antibodies or cytokines (Figure
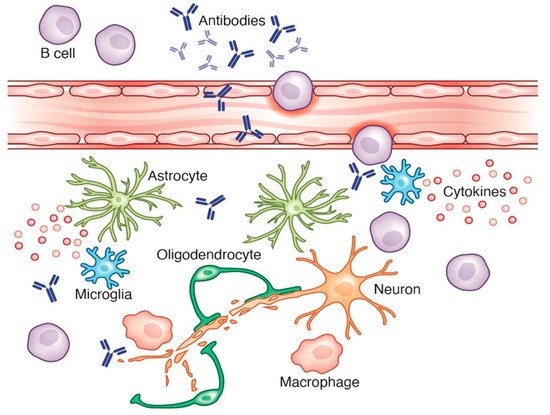
2).
Figure 2. Activated B cells can produce antibodies in an inflammatory setting. Antibodies are capable of activating the complement system, which can activate microglia and astrocytes to release pro-inflammatory factors, leading to neuronal loss. Credit: Katie Vicari.
2.3. Alzheimer’s Disease
Alzheimer’s disease (AD) is a progressive neurologic disorder that results in a continuous decline in behavioral, social, and cognitive function as a result of brain atrophy. Alzheimer’s disease pathology is characterized by the presence of ß-amyloid (Aß) plaques
[57] and neurofibrillary tangles (NFT)
[58]. Despite extensive studies examining the generation, molecular composition, and potential effects of Aß plaques and NFTs in AD, there is no effective treatment for the eventual cognitive decline. In recent years, AD has increasingly been recognized as a neurodegenerative disease characterized by dysregulation of the CNS due to innate and adaptive immune responses. In
post-mortem AD samples, significant inflammation in the CNS has been observed, mainly by positron emission tomography (PET) targeting microglial activation and reactive astrocyte activity
[59][60][61]. Similar to MS pathophysiology, it is increasingly evident that patients with AD exhibit a sustained immune response, possibly due to a disruption of the BBB
[62][63]. Deposits of Aß in the CNS vasculature increase cytotoxicity and CNS inflammation that can further disrupt BBB permeability and exacerbate disease
[64][65] by allowing cells and molecules from the periphery into the CNS, potentially activating microglia and astrocyte populations to an inflammatory phenotype. This immunological component of AD is further supported by a recent study on AD in mice that showed an improvement in disease symptoms after depletion of B cells or genetic loss of B cells
[66]. Additionally, the B cells in AD mice acquired an inflammatory phenotype as shown by the upregulation of pro-inflammatory cytokines, including IL-6, TNFα, and IFNγ. The sustained immune response in AD brains as well as recent animal studies suggesting pathogenic B cells in AD provide compelling evidence of a role for B cells in neuroinflammation and AD pathophysiology.
 +1 point
+1 point