In recent years, the role of B cells in neurological disorders has substantially expanded our perspectives on mechanisms of neuroinflammation. The success of B cell-depleting therapies in patients with CNS diseases such as neuromyelitis optica and multiple sclerosis has highlighted the importance of neuroimmune crosstalk in inflammatory processes. While B cells are essential for the adaptive immune system and antibody production, they are also major contributors of pro- and anti-inflammatory cytokine responses in a number of inflammatory diseases. B cells can contribute to neurological diseases through peripheral immune mechanisms, including production of cytokines and antibodies, or through CNS mechanisms following compartmentalization. Emerging evidence suggests that aberrant pro- or anti-inflammatory B cell populations contribute to neurological processes, including glial activation, which has been implicated in the pathogenesis of several neurodegenerative diseases.
- B cell
- neuroinflammation
- neurological disorders
- cytokines
- multiple sclerosis
- Parkinson’s disease
- Alzheimer’s disease
1. Introduction
2. B Cells in Neurological Diseases
2.1. Multiple Sclerosis
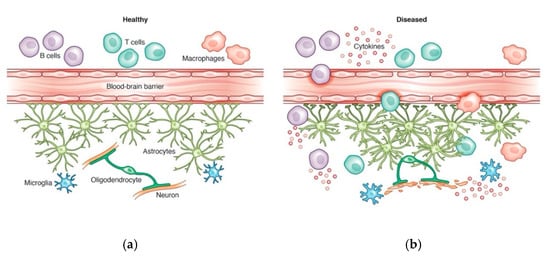
2.2. Parkinson’s Disease
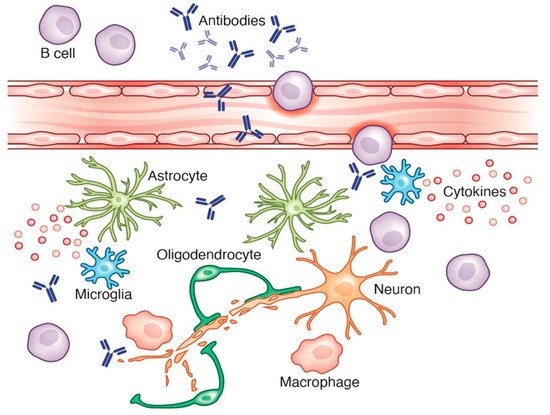
2.3. Alzheimer’s Disease
This entry is adapted from the peer-reviewed paper 10.3390/cells10071605
References
- Machado-Santos, J.; Saji, E.; Tröscher, A.R.; Paunovic, M.; Liblau, R.; Gabriely, G.; Bien, C.G.; Bauer, J.; Lassmann, H. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 2018, 141, 2066–2082.
- Pardridge, W.M.; Boado, R.J.; Patrick, D.J.; Hui, E.K.-W.; Lu, J.Z. Blood-Brain Barrier Transport, Plasma Pharmacokinetics, and Neuropathology Following Chronic Treatment of the Rhesus Monkey with a Brain Penetrating Humanized Monoclonal Antibody Against the Human Transferrin Receptor. Mol. Pharm. 2018, 15, 5207–5216.
- Pinheiro, M.A.L.; Kooij, G.; Mizee, M.R.; Kamermans, A.; Enzmann, G.; Lyck, R.; Schwaninger, M.; Engelhardt, B.; de Vries, H.E. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 461–471.
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.; Rouhani, S.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nat. Cell Biol. 2015, 523, 337–341.
- Harling-Berg, C.; Knopf, P.M.; Merriam, J.; Cserr, H.F. Role of cervical lymph nodes in the systemic humoral immune response to human serum albumin microinfused into rat cerebrospinal fluid. J. Neuroimmunol. 1989, 25, 185–193.
- Widner, H.; Möller, G.; Johansson, B.B. Immune Response in Deep Cervical Lymph Nodes and Spleen in the Mouse after Antigen Deposition in Different Intracerebral Sites. Scand. J. Immunol. 1988, 28, 563–571.
- Harling-Berg, C.J.; Park, J.T.; Knopf, P.M. Role of the cervical lymphatics in the Th2-type hierarchy of CNS immune regulation. J. Neuroimmunol. 1999, 101, 111–127.
- Stern, J.N.; Yaari, G.; Heiden, J.V.; Church, G.; Donahue, W.F.; Hintzen, R.; Huttner, A.J.; Laman, J.; Nagra, R.M.; Nylander, A.; et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med. 2014, 6, 248ra107.
- Palanichamy, A.; Apeltsin, L.; Kuo, T.C.; Sirota, M.; Wang, S.; Pitts, S.J.; Sundar, P.D.; Telman, D.; Zhao, L.Z.; Derstine, M.; et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci. Transl. Med. 2014, 6, 248ra106.
- Kowarik, M.C.; Grummel, V.; Wemlinger, S.; Buck, D.; Weber, M.S.; Berthele, A.; Hemmer, B. Immune cell subtyping in the cerebrospinal fluid of patients with neurological diseases. J. Neurol. 2014, 261, 130–143.
- Magliozzi, R.; Howell, O.; Nicholas, R.; Cruciani, C.; Castellaro, M.; Romualdi, C.; Rossi, S.; Pitteri, M.; Benedetti, M.D.; Gajofatto, A.; et al. Inflammatory intrathecal profiles and cortical damage in multiple sclerosis. Ann. Neurol. 2018, 83, 739–755.
- Inglese, M.; Oesingmann, N.; Casaccia, P.; Fleysher, L. Progressive Multiple Sclerosis and Gray Matter Pathology: An MRI Perspective. Mt. Sinai J. Med. 2011, 78, 258–267.
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116.
- Baroncini, D.; Ghezzi, A.; Annovazzi, P.; Colombo, B.; Martinelli, V.; Minonzio, G.; Moiola, L.; Rodegher, M.; Zaffaroni, M.; Comi, G. Natalizumab versus fingolimod in patients with relapsing-remitting multiple sclerosis non-responding to first-line injectable therapies. Mult. Scler. J. 2016, 22, 1315–1326.
- Frischer, J.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721.
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2006, 130, 1089–1104.
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; De Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220.
- Elliott, C.; Wolinsky, J.S.; Hauser, S.L.; Kappos, L.; Barkhof, F.; Bernasconi, C.; Wei, W.; Belachew, S.; Arnold, D.L. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Mult. Scler. J. 2018, 25, 1915–1925.
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219.
- Patsopoulos, N.A.; Baranzini, S.E.; Santaniello, A.; Shoostari, P.; Cotsapas, C.; Wong, G. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365.
- International Multiple Sclerosis Genetics Consortium. A systems biology approach uncovers cell-specific gene regulatory effects of genetic associations in multiple sclerosis. Nat. Commun. 2019, 10.
- Hussman, J.; Beecham, A.H.; Schmidt, M.; Martin, E.R.; McCauley, J.L.; Vance, J.; Haines, J.L.; Pericak-Vance, M. GWAS analysis implicates NF-κB-mediated induction of inflammatory T cells in multiple sclerosis. Genes Immun. 2016, 17, 305–312.
- Weber, M.S.; Prod’Homme, T.; Patarroyo, J.C.; Molnarfi, N.; Karnezis, T.; Lehmann-Horn, K.; Danilenko, D.M.; Eastham-Anderson, J.E.; Slavin, A.J.; Linington, C.; et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann. Neurol. 2010, 68, 369–383.
- Hart, B.A.; Dunham, J.; Faber, B.W.; Laman, J.D.; Van Horssen, J.; Bauer, J.; Kap, Y.S. A B Cell-Driven Autoimmune Pathway Leading to Pathological Hallmarks of Progressive Multiple Sclerosis in the Marmoset Experimental Autoimmune Encephalomyelitis Model. Front. Immunol. 2017, 8, 804.
- Häusler, D.; Häusser-Kinzel, S.; Feldmann, L.; Torke, S.; Lepennetier, G.; Bernard, C.C.A.; Zamvil, S.S.; Brück, W.; Lehmann-Horn, K.; Weber, M.S. Functional characterization of reappearing B cells after anti-CD20 treatment of CNS autoimmune disease. Proc. Natl. Acad. Sci. USA 2018, 115, 9773–9778.
- Smolders, J.; Heutinck, K.M.; Fransen, N.L.; Remmerswaal, E.B.M.; Hombrink, P.; Berge, I.J.M.T.; Van Lier, R.A.W.; Huitinga, I.; Hamann, J. Tissue-resident memory T cells populate the human brain. Nat. Commun. 2018, 9.
- McPherson, R.C.; Cambrook, H.E.; O’Connor, R.A.; Anderton, S.M. Induction of Passive EAE Using Myelin-Reactive CD4+ T Cells. Methods Mol. Biol. 2014, 1193, 187–198.
- Maimaitijiang, G.; Watanabe, M.; Shinoda, K.; Isobe, N.; Nakamura, Y.; Masaki, K.; Matsushita, T.; Yoshikai, Y.; Kira, J.-I. Long-term use of interferon-β in multiple sclerosis increases Vδ1−Vδ2−Vγ9− γδ T cells that are associated with a better outcome. J. Neuroinflamm. 2019, 16, 1–15.
- Scheu, S.; Ali, S.; Mann-Nüttel, R.; Richter, L.; Arolt, V.; Dannlowski, U.; Kuhlmann, T.; Klotz, L.; Alferink, J. Interferon β-Mediated Protective Functions of Microglia in Central Nervous System Autoimmunity. Int. J. Mol. Sci. 2019, 20, 190.
- Kap, Y.S.; Bauer, J.; Van Driel, N.; Bleeker, W.K.; Parren, P.W.; Kooi, E.-J.; Geurts, J.J.; Laman, J.D.; Craigen, J.L.; Blezer, E.; et al. B-Cell Depletion Attenuates White and Gray Matter Pathology in Marmoset Experimental Autoimmune Encephalomyelitis. J. Neuropathol. Exp. Neurol. 2011, 70, 992–1005.
- Kap, Y.S.; Van Driel, N.; Blezer, E.; Parren, P.W.H.I.; Bleeker, W.K.; Laman, J.D.; Craigen, J.L.; Hart, B.A. ’T Late B Cell Depletion with a Human Anti-Human CD20 IgG1κ Monoclonal Antibody Halts the Development of Experimental Autoimmune Encephalomyelitis in Marmosets. J. Immunol. 2010, 185, 3990–4003.
- Obermeier, B.; Mentele, R.; Malotka, J.; Kellermann, J.; Kümpfel, T.; Wekerle, H.; Lottspeich, F.; Hohlfeld, R.; Dornmair, K. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat. Med. 2008, 14, 688–693.
- Barnett, M.H.; Parratt, J.D.E.; Cho, E.-S.; Prineas, J.W. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Ann. Neurol. 2009, 65, 32–46.
- Brändle, S.M.; Obermeier, B.; Senel, M.; Bruder, J.; Mentele, R.; Khademi, M.; Olsson, T.; Tumani, H.; Kristoferitsch, W.; Lottspeich, F.; et al. Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self-proteins. Proc. Natl. Acad. Sci. USA 2016, 113, 7864–7869.
- Petzold, A. Intrathecal oligoclonal IgG synthesis in multiple sclerosis. J. Neuroimmunol. 2013, 1–10.
- Freedman, M.S.; Thompson, E.J.; Deisenhammer, F.; Giovannoni, G.; Grimsley, G.; Keir, G.; Öhman, S.; Racke, M.K.; Sharief, M.; Sindic, C.J.M.; et al. Recommended Standard of Cerebrospinal Fluid Analysis in the Diagnosis of Multiple Sclerosis: A consensus statement. Arch. Neurol. 2005, 865–870.
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-Cell Depletion with Rituximab in Relapsing–Remitting Multiple Sclerosis. N. Engl. J. Med. 2008, 358, 676–688.
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471.
- Lu, T.; Shu, Y.; Dai, Y.; Liu, X.; Chang, Y.; Huang, Q.; Kermode, A.G.; Qiu, W. B cell depleting therapy for multiple sclerosis overlapping with neuromyelitis optica spectrum disorder. Mult. Scler. Relat. Disord. 2018, 22, 83–85.
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 898–915.
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s Disease: Substantia Nigra Regional Selectivity. Brain 1991, 114, 2283–2301.
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain: II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122, 1437–1448.
- Papachroni, K.K.; Ninkina, N.; Papapanagiotou, A.; Hadjigeorgiou, G.M.; Xiromerisiou, G.; Papadimitriou, A.; Kalofoutis, A.; Buchman, V.L. Autoantibodies to alpha-synuclein in inherited Parkinson’s disease. J. Neurochem. 2006, 101, 749–756.
- Bas, J.; Calopa, M.; Mestre, M.; Molleví, D.G.; Cutillas, B.; Ambrosio, S. Lymphocyte populations in Parkinson’s disease and in rat models of parkinsonism. J. Neuroimmunol. 2001, 113, 146–152.
- Orr, C.; Rowe, D.; Mizuno, Y.; Mori, H.; Halliday, G.M. A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 2005, 128, 2665–2674.
- Pienaar, I.S.; Lee, C.H.; Elson, J.L.; McGuinness, L.; Gentleman, S.M.; Kalaria, R.N.; Dexter, D.T. Deep-brain stimulation associates with improved microvascular integrity in the subthalamic nucleus in Parkinson’s disease. Neurobiol. Dis. 2015, 74, 392–405.
- Gray, M.T.; Woulfe, J.M. Striatal Blood–Brain Barrier Permeability in Parkinson’S Disease. Br. J. Pharmacol. 2015, 35, 747–750.
- Wong, K.; Grove, J.; Grandinetti, A.; Curb, J.; Yee, M.; Blanchette, P.; Ross, G.; Rodriguez, B. Association of Fibrinogen with Parkinson Disease in Elderly Japanese-American Men: A Prospective Study. Neuroepidemiology 2010, 34, 50–54.
- Raghunathan, R.; Hogan, J.D.; Labadorf, A.; Myers, R.H.; Zaia, J. A glycomics and proteomics study of aging and Parkinson’s disease in human brain. Sci. Rep. 2020, 10, 12804.
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2008, 119, 182–192.
- Besong-Agbo, D.; Wolf, E.; Jessen, F.; Oechsner, M.; Hametner, E.; Poewe, W.; Reindl, M.; Oertel, W.H.; Noelker, C.; Bacher, M.; et al. Naturally occurring -synuclein autoantibody levels are lower in patients with Parkinson disease. Neurology 2012, 80, 169–175.
- Han, M.; Nagele, E.; DeMarshall, C.; Acharya, N.; Nagele, R. Diagnosis of Parkinson’s Disease Based on Disease-Specific Autoantibody Profiles in Human Sera. PLoS ONE 2012, 7, e32383.
- He, Y.; Le, W.-D.; Appel, S.H. Role of Fcγ Receptors in Nigral Cell Injury Induced by Parkinson Disease Immunoglobulin Injection into Mouse Substantia Nigra. Exp. Neurol. 2002, 176, 322–327.
- Stevens, C.; Rowe, D.; Morel-Kopp, M.-C.; Orr, C.; Russell, T.; Ranola, M.; Ward, C.; Halliday, G. Reduced T helper and B lymphocytes in Parkinson’s disease. J. Neuroimmunol. 2012, 252, 95–99.
- Souto-Carneiro, M.M.; Mahadevan, V.; Takada, K.; Fritsch-Stork, R.; Nanki, T.; Brown, M.; Fleisher, T.; Wilson, M.; Goldbach-Mansky, R.; Lipsky, P. Alterations in peripheral blood memory B cells in patients with active rheumatoid arthritis are dependent on the action of tumour necrosis factor. Arthritis Res. Ther. 2009, 11, R84.
- Hansen, A.; Odendahl, M.; Reiter, K.; Jacobi, A.M.; Feist, E.; Scholze, J.; Burmester, G.R.; Lipsky, P.E.; Dorner, T. Diminished peripheral blood memory B cells and accumulation of memory B cells in the salivary glands of patients with Sjögren’s syndrome. Arthritis Rheum. 2002, 46, 2160–2171.
- Selkoe, D.J. Normal and Abnormal Biology of the β-Amyloid Precursor Protein. Annu. Rev. Neurosci. 1994, 17, 489–517.
- Alonso, A.D.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787.
- Knezevic, D.; Mizrahi, R. Molecular imaging of neuroinflammation in Alzheimer’s disease and mild cognitive impairment. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2018, 80, 123–131.
- Zimmer, E.R.; Leuzy, A.; Benedet, A.L.; Breitner, J.; Gauthier, S.; Rosa-Neto, P. Tracking neuroinflammation in Alzheimer’s disease: The role of positron emission tomography imaging. J. Neuroinflamm. 2014, 11, 120.
- Lagarde, J.; Sarazin, M.; Bottlaender, M. In vivo PET imaging of neuroinflammation in Alzheimer’s disease. J. Neural Transm. 2017, 125, 847–867.
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.D.; Zlokovic, B.V. Deficiency in Mural Vascular Cells Coincides with Blood-Brain Barrier Disruption in Alzheimer’s Disease. Brain Pathol. 2013, 23, 303–310.
- Ryu, J.K.; McLarnon, J.G. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J. Cell. Mol. Med. 2009, 13, 2911–2925.
- Erickson, M.A.; Banks, W.A. Blood–Brain Barrier Dysfunction as a Cause and Consequence of Alzheimer’s Disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513.
- Carrano, A.; Hoozemans, J.J.; Van Der Vies, S.M.; Rozemuller, A.J.; Van Horssen, J.; De Vries, H.E. Amyloid Beta Induces Oxidative Stress-Mediated Blood–Brain Barrier Changes in Capillary Amyloid Angiopathy. Antioxid. Redox Signal. 2011, 15, 1167–1178.
- Kim, K.; Wang, X.; Ragonnaud, E.; Bodogai, M.; Illouz, T.; DeLuca, M.; McDevitt, R.A.; Gusev, F.; Okun, E.; Rogaev, E.; et al. Therapeutic B-cell depletion reverses progression of Alzheimer’s disease. Nat. Commun. 2021, 12, 1–11.