 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Thomas Dittmar | + 3666 word(s) | 3666 | 2021-06-16 07:56:24 | | | |
| 2 | Conner Chen | Meta information modification | 3666 | 2021-06-28 09:47:39 | | |
Video Upload Options
In addition to physiological processes, such as fertilization, placentation, myogenesis, osteoclastogenesis and wound healing/tissue regeneration the biological phenomenon of cell-cell fusion also plays a role in cancer, which was already postulated by the German physician Otto Aichel in 1911. Indeed, cancer cells could either fuse with other cancer cells or could hybridize with macrophages, fibroblasts and stem cells, thereby giving rise to tumor hybrid cells that could exhibit novel properties, such as an increased drug resistance and/or an enhanced metastasis formation capacity.
1. How Do Cancer Cells Fuse?
Even though it is known that cancer cells, like other cells, possess fusogenic capacities, it still remains to be elucidated how cell–cell fusion is directed. First of all, cell-cell fusion is a complex and tightly regulated process. Cells are not per se fusogenic, but have to enter a pro-fusogenic state first [1][2][3][4][5][6]. Likewise, hybridized cells have to return into a non-fusogenic state to prevent further uncontrolled fusion events.
It is well known that cell-cell fusion is facilitated by so-called fusogens, which bring the lipid bilayers of two cells into immediate contact, catalyze the formation of energy-intensive fusion intermediates and open the fusion pore [5]. However, only a handful of fusogens have been identified so far in humans, which usually direct physiological cell–cell fusion processes, such as syncytins in placentation, myomaker and myomerger in myogenesis, and IZUMO and Juno in fertilization [7][8][9][10][11][12][13][14]. Several studies revealed that syncytins were also involved in tumor cell-cell fusion events [15][16][17][18]. For instance, Bjerregard and colleagues demonstrated that breast cancer endothelial cell fusion events were mediated by syncytin-1 [15], which, surprisingly, was associated with a better prognosis for afflicted patients [19]. In contrast, the overexpression of syncytin-1 in urothelial cell carcinoma of the bladder has been proposed as an indicator for urothelial cell carcinoma risk since it was associated with proliferation, viability and an increased fusion frequency [18]. Likewise, proliferation and cell–cell fusion of endometrial carcinoma were induced by syncytin-1 [16]. Moreover, the authors further demonstrated that the syncytin-1-mediated fusion of endometrial cancer cells was inversely correlated to transforming growth factor-β (TGF-β) levels. Thereby, high TGF-β levels inhibited cell–cell fusion, but induced cell proliferation, whereas blocking of TGF-β was associated with an enhanced frequency of cell–cell fusion events [16]. The finding that the cell–cell fusion of endometrial cancer cells is regulated by TGF-β is in agreement to the hypothesis that cell-cell fusion is a tightly controlled process. In this connection, Yan and colleagues also showed that the Wnt/β-catenin-mediated up-regulation of syncytin-1 expression contributed to the tumor necrosis factor-α (TNF-α)-enhanced fusion of human umbilical vein endothelial cells (HUVECs) and oral squamous cell carcinoma (OSCC) cells [17]. While these data further substantiate the idea that cell-cell fusion is a tightly regulated process, they also indicate that cell-cell fusion could be induced. Indeed, TNF-α or inflammatory conditions in general have been associated with an enhanced cell-cell fusion frequency [20][21][22][23][24][25][26][27]. However, it remains to be elucidated how TNF-α or inflammatory conditions ultimately induce cell-cell fusion and/or support/induce the conversion of cells from a non-fusogenic into a fusogenic state. In this connection, we have recently demonstrated that the TNF-α induced fusion of human M13SV1-Cre breast epithelial cells with human MDA-MB-435-pFDR1 cancer cells was attributed to matrix metalloproteinase 9 (MMP9) expression [25]. Both the blockade of MMP9 activity using a specific inhibitor [25] and the inhibition of MMP9 expression by minocycline [24] markedly impaired the TNF-α-induced cell-cell fusion rate. Interestingly, a similar mechanism has also been described for macrophages [26], but it remains completely unclear how MMP9 is involved in the entire cell-cell fusion process. It might be possible that proteolytic enzymes such as MMPs or ADAMs may reduce the overall distance between both fusion partners by degrading cell adhesion molecules, mobilize and activate masked growth factors embedded in the extracellular matrix or facilitating cell-cell fusion by fostering cell–cell interactions [28][29][30]. In addition to MMP9 or proteases in general, several other proteins/molecules, such as cell adhesion molecules, cytokines, chemokines, receptors, actin-modifying enzymes and lipids have been identified that are all involved in cell–cell fusion (for review see: [1][2][3][4][31][5]), which substantiate the complexity of this process.
It is well known that the tumor microenvironment resembles chronically inflamed tissue [32][33][34] and because of that, tumors have been proposed as wounds that do not heal [35]. As inflammation/inflammatory conditions induce cell-cell fusion, it might be speculated that the chronically inflamed tumor microenvironment provides a fusion-friendly milieu. In any case, the number of cell–cell fusion events within the tumor microenvironment remains ambiguous. Indeed, animal studies have shown that the intratumoral cancer cell–cell fusion frequency could be between 0.0066% to 6% [36][37][38][39][40], but these estimated counts are only valid for those tumor hybrids that have been detected due to the co-expression of specific fusion markers. Homotypic tumor cell-cell fusion events or tumor hybrids that have lost the expression of the fusion markers will not be considered as hybrids in the evaluation. Such indistinguishable or invisible tumor hybrids have also been referred to as dark matter hybrids, which could contribute to tumor growth and progression; albeit they cannot yet be detected and quantified [41]. Likewise, cancer cell lines differed markedly between each other regarding their fusogenic capacities [36][42], indicating that highly fusogenic and less fusogenic cancer cell lines exist, which could also have an impact on the frequency of cell–cell fusion events. Similar findings were also presented for cells of the hematopoietic lineage, such as macrophages, B-lymphocytes and T-lymphocytes, which were all capable of fusing with transformed intestinal cells [43]. However, in comparison to macrophages, which were the main fusion partners (about 20%) the fusogenic capacity of B- and T-lymphocytes was rather low (about 3%) [43]. Moreover, the frequency of cell–cell fusion events detected in vivo likely depends on additional parameters, such as the used cancer cell lines (human vs. murine origin), the site of implantation (orthotropically vs. non-orthotropically), the addition of matrix components and cells (which have an impact on tumor cell engraftment) and the used mouse strain (immune compromised mice strains: BALB/c nude, non-obese diabetic/severe combined immunodeficiency (NOD/SCID) vs. non-immunocompromised (transgenic) mice strains). All these parameters could have an impact on the total number of cell–cell fusion events in animal tumor models. A small tumor size consisting of less fusogenic cancer cells would result in a low frequency of cell–cell fusion, whereas the rate of tumor hybrids would be higher in large tumors comprising of highly fusogenic cancer cells. Immunocompromised mice strains are routinely used for tumorigenic studies using human cancer cell lines, but they lack B- and T-lymphocytes, which exhibit a certain degree of fusogenecity. It is known that human cancer cells could fuse with murine macrophages and stromal cells [44][45][46], but it remains unclear whether the frequency of human × mouse cell–cell fusion events is comparable to the frequency of human cell–cell fusion events.
Whether the cell–cell fusion frequency in human cancers could be as high as observed in animal studies remains to be elucidated. As stated above, only visible tumor hybrid cells can be detected so far, whereas invisible tumor hybrids remain undetected. Likewise, the total rate of cell–cell fusion events depends on several parameters, such as the overall fusogenecity of cancer cells and normal cells and cell-cell fusion influencing factors/conditions, which could vary between different types of cancer. Moreover, the cell–cell fusion frequency number only indicates the rate of cell–cell fusion events that have been determined at a certain time point, but it neither represents the total number of surviving tumor hybrids nor whether tumor hybrids possess novel properties. Both depend on the outcome of post-fusion processes.
2. Post-Fusion Processes
Most studies indicate that tumor hybrids are more aggressive and more malignant than their parental cancer cells [36][47][42][48][49][50][37][51][43][52][53][54][55][56][57][58][59][44][60][61][62][45][63][64][65][66][67], suggesting that cell–cell fusion might be a directed process which per se converts low-malignant tumor cells into highly malignant cancer cells through hybridization with other cells. This suggestion, however, is not correct, since the ultimate phenotype of tumor hybrids cannot be predicted, which is attributed to post-fusion processes, such as HST/PR [68][69][70] and PHSP/AKE [57][71][72] that run in a unique manner in each evolved tumor hybrid cell.
Cell fusion first results in bi- (or multi-) nucleated cells, also termed heterokaryons, which could either remain in this state or could undergo HST/PR [68][69][70], thereby giving rise to daughter cells with one-half chromosomal content. In fact, HST/PR is a complex and not yet fully understood process. HST/PR requires an active cell cycle and the resolution of both nuclear membranes results in the random merging of parental chromosomes, which are subsequently segregated via tripolar and multipolar divisions, thereby giving rise to aneuploid daughter cells [69][70] (Figure 1).
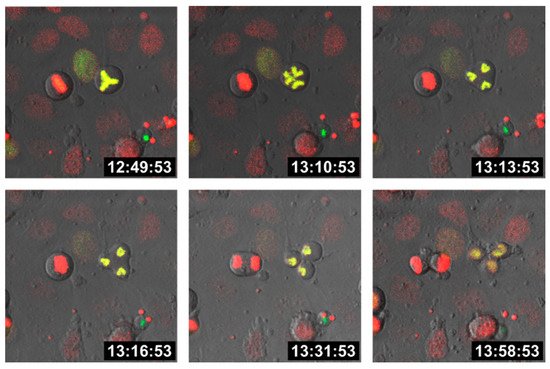
Figure 1. Tripolar division of a hybrid cell. M13SV1 human breast epithelial cells [73] were either stably transduced with pH2B-GFP (kind gift from Geoff Wahl; Addgene plasmid #11680; http://n2t.net/addgene:11680; RRID: Addgene_11680; accessed on 15 April 2021) or pH2B_mCherry_IRES_puro2 (kind gift from Daniel Gerlich; Addgene plasmid #21045; http://n2t.net/addgene:21045; accessed on 15 April 2021, RRID:Addgene_21045). Hybrid cells were cultured on chamber slides (ThermoFisher Scientific GmbH, Schwerte, Germany) and time-lapse series were recorded using a Leica TCS SP5 confocal laser scanning microscope (Leica, Wetzlar, Germany).
Moreover, HST/PR is further associated with additional chromosomal aberrations, such as double strand breaks and chromothripsis [69][70][74][75]. Indeed, chromosome missegregation, particularly when coupled with breakage, is often followed by irreversible cell cycle arrest, impaired proliferation or cell death [76]. This could either be attributed to the lack of crucial chromosomes or due to the induction of apoptosis if tumor cells have fused with normal cells harboring a functional set of tumor suppressor genes. This is in view, findings suggest that breast cancer cell × cancer-associated fibroblast hybrids were not viable, whereas homotypic and heterotypic breast cancer cell hybrids could be generated and propagated [36].
In addition to these initial HST/PR dependent events, the genomic background of tumor hybrids is further fine-tuned during the next divisions to give rise to stable proliferating tumor hybrids. This fine-tuning process, which has also been named the post-hybrid selection process (PHSP) [57][71], resembles to the so-called autocatalytic karyotypic evolution (AKE) model that was proposed for aneuploid cells [72] and which is associated with additional gains and losses of whole chromosomes, further chromosomal aberrations and an overall increased genomic/epigenomic instability (Figure 2).
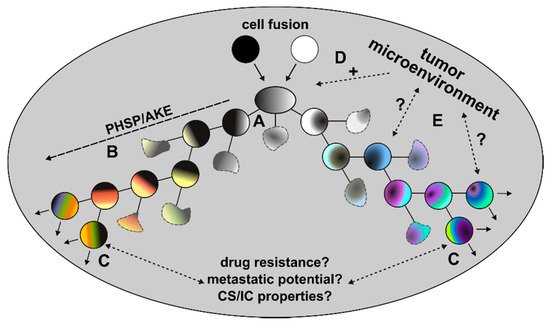
Figure 2. Post-fusion processes. Due to the random segregation of merged parental chromosomes, different viable and non-viable tumor hybrid cells with a unique karyotype originate (A). Surviving tumor hybrids will first undergo PHSP/AKE, which is a fitting process that gives rise to tumor hybrids with a rather stable, but still aneuploid and genomic instable karyotype (B). The initial period of PHSP/AKE might be associated with an increased cell death (blurry cells). Surviving and proliferating tumor hybrids could exhibit novel characteristics, such as an increased drug resistance, metastatic potential or CS/IC properties (C). The role of the tumor microenvironment in these processes remains unclear. While it is assumed that the chronically inflamed tumor microenvironment might positively trigger cell-cell fusion events (D), its role in PHSP/AKE and acquisition of novel properties of tumor hybrids remains unclear (E).
Like HST/PR, PHSP/AKE also runs in a unique manner in each tumor hybrid cell, thereby giving rise to unique progenies. For instance, Zhao and colleagues demonstrated that the number of chromosomes in fusion-derived clones near triploid or tetraploid at early passage usually decreased with repeated passage [77]. Likewise, a substantial shift in the phenotypes of MDA-MB-231 × SUM159PT hybrids at early (2) and extended (10) passages was observed, which was further consistent with the selection of a fit subpopulation of hybrid cells and additional diversification [36]. Hybrid clones derived from human breast epithelial cells and human breast cancer cells possessed a unique mean chromosomal number [78][79], which is in accordance with recent data of Delespaul et al. and Lartigue and colleagues who also showed that mesenchymal tumor hybrid clones exhibited a unique chromosomal content [80][81][82]. In this connection, it should be kept in mind that PSHP/AKE does not stop at a certain time point, e.g., after 10 or 15 divisions. It is a continuous process, whereby it might be speculated that it runs much faster during early passages and much slower during late passages once a relative stable karyotype has been established in tumor hybrids. Nonetheless, aneuploid/genomic/epigenomic instable tumor hybrid cells will always give rise to aneuploid/genomic/epigenomic instable daughter hybrids, etc., which is definitely associated with an increase in intratumoral heterogeneity.
In vitro studies revealed that the incidence of tumor hybrids surviving both HST/PR and PHSP/AKE could be up to 1% [42][83]. How many tumor hybrids will survive both HST/PR and PHSP/AKE in vivo remains unclear. As mentioned above, only those cells possessing specific fusion markers are considered as tumor hybrids, which excludes homotypic tumor hybrids and cells that have lost marker expression. Likewise, it cannot be ruled out that the specific survival rates of discrete tumor hybrid populations might differ, which would be similar to the unique fusogenic properties of cancer cells and normal cells. For instance, no colonies were derived from 1456 tracked RL-1/HPS-14 cancer–stromal hybrids, whereas 41 colonies were grown from 1456 tracked RL-1/HPS-15 cancer–stromal hybrids [84]. HPS-14 cells represent normal human prostate myofibroblast stromal cells, whereas HPS-15 cells are cancer-associated human prostate myofibroblast stromal cells [84]. Due to the low tumor hybrid survival rate, the authors speculated whether the fusion of cancer cells with stromal cells would be suitable for cancer therapy since the primary fate of cancer stromal hybrids was death [84]. Likewise, different numbers of tumor hybrid clones were derived from human M13SV1-EGFP-Neo breast epithelial cells and human HS578T-Hyg breast cancer cells (M13HS-X; X = 1–5, 7–10), human MDA-MB-435-Hyg cancer cells (M13MDA435-X, X = 1–4) and human MDA-MB-231-Hyg breast cancer cells (M13MDA231-X, X = 1–14) [50][79]. Given that identical cell numbers have been used for co-culture experiments (1 × 106 cells per cell line) these data indicate that the overall survival rate of tumor hybrids in relation to the total number of parental cells was rather low [50][79].
However, it has to be considered that cell-cell fusion is not a one-time event, but rather a continuous process, suggesting that the total number of tumor hybrids should increase steadily with time. Likewise, tumor hybrids are proliferating and give rise to progenies, which will also steadily increase the pool of tumor hybrids within the tumor microenvironment. Moreover, all these calculations and considerations are based on visible tumor hybrids, whereas the unrecognized fraction of invisible tumor hybrids is excluded, suggesting that both the fusion frequency and survival rate could be higher than anticipated. Mathematical models of Miroshnychenko and colleagues demonstrated that the intratumoral heterogeneity is much more driven by both fusion and mutations than mutations alone despite rather low fusion probabilities of 6.6 × 10−3 in vitro and 6.6 × 10−5 in vivo [36]. Interestingly, higher levels of mutational heterogeneity dramatically enhanced the impact of fusion-mediated recombination, which should therefore be more pronounced in tumors with higher levels of mutational clonal heterogeneity [36]. Moreover, cell-cell fusion-mediated diversity was acquired much faster and higher in 3D than in 2D contexts, which is likely attributed to the higher number of genetically distinct neighbors [36]. Hence, even a rather low fusion (and survival) probability could have a huge impact on intratumoral heterogeneity and tumor progression.
3. Novel Properties of Tumor Hybrids
Cell fusion is an open process, which means that it could not be predicted whether tumor cells will be viable or non-viable and whether they could exhibit novel properties or not. Hence, the fraction of viable tumor hybrids will be a mixture of diverse hybrid clones exhibiting individual properties, like a rainbow consisting of different colors. Some hybrid clones could be more sensitive to cancer therapy, whereas other hybrid clones could be highly drug-resistant. Likewise, some hybrids could be non-metastatic, whereas other hybrid clones could be highly metastatic. It is also possible that individual hybrid clones could be both more metastatic and drug-resistant, whereas other hybrids are either metastatic or drug-resistant, and some could be neither metastatic nor drug-resistant. Finally, some tumor hybrid clones could be highly proliferative, whereas others rather remain in a quiescent state. In fact, some studies demonstrated that cell-cell fusion could also give rise to tumor hybrid cells exhibiting a weaker or even no metastatic capacity as compared to the parental cancer cell line. This, for instance, was demonstrated by Rachkovsky and colleagues [44]. While the majority of macrophage × melanoma hybrids were more aggressive and produced metastases sooner and in more mice, the metastatic capacity of some tumor hybrids was lower or even zero in comparison to the parental cancer cell line [44]. Fahlbusch et al. have recently shown that hybrid cells that were derived from human breast epithelial cells exhibiting stem cell properties and human breast cancer cells varied markedly regarding their capacities to form mammospheres and the amount of ALDH1-positive cells [79]. Tumor hybrids that were derived from murine neu+ mammary cancer and mouse macrophages exhibited no increased metastatic potential [59].
How many more malignant tumor hybrid clones will evolve in comparison to all tumor hybrid clones remains ambiguous. Nonetheless, it can be assumed that the probability that surviving tumor hybrid clones with novel properties will originate is directly correlated to the total number of evolved tumor hybrid clones. This assumption is supported by mathematical data revealing that despite a low fusion rate, the diversity and clonal richness is much more driven by both fusion and mutations [36]. Moreover, the impact of cell-cell fusion on the diversification of individual tumor hybrid clones was substantially higher in a 3D environment due to higher numbers of genetically distinct neighbor cells [36], suggesting that tumor hybrids exhibiting novel properties might originate from heterotypic rather than from homotypic cell–cell fusion events.
These data further support the importance of the chronically inflamed tumor microenvironment, which may not only provide a fusion-friendly milieu, but also provides a high number of different fusion partners. The chronically inflamed tumor microenvironment represents a complex mixture of tumor cells, fibroblasts, immune cells, MSCs, endothelial cells, neurons, extracellular matrix components, cytokines, chemokines, growth factors, proteases, hormones and neurotransmitters, which all physically and chemically interact with each other [71][85][86][87]. Likewise, the chronically inflamed tumor microenvironment plays a crucial role in directing various processes, such as EMT, trans- or retrodifferentiation, autophagy, metastases formation and epigenetic alterations [71][85][86][87] and putatively even PHSP [71]. Whether the chronically inflamed tumor microenvironment might provide survival signals ensuring that tumor hybrids will successfully pass through this karyotype fine-tuning process or whether it might have an impact on the final phenotype of tumor hybrids is not clear but cannot be ruled out. The chronically inflamed tumor microenvironment consists of different spatial habitats, each harboring a specific set of cellular and environmental conditions, which all closely interact with and influence each other [88][89][90]. Hence, the cellular composition has an impact on the composition of the environment and vice versa, which additionally drives intratumoral heterogeneity in a Darwinian manner [88][89][90]. With regard to cell–cell fusion, this would mean that the specific spatial habitat in which tumor hybrids have formed could also have an impact on the cells’ phenotype, since evolving tumor hybrids have to adopt to the specific spatial environment in order to survive. It is commonly accepted that CS/ICs reside in a specialized compartment of the tumor microenvironment termed niche [91]. Fusion between cancer cells and MSCs or stem-like cells could give rise to tumor hybrid cells exhibiting a CS/IC phenotype [50][54][92][20][38][93][94][78], suggesting that the origin of tumor hybrids close to this specialized compartment might give rise to CS/IC-like tumor hybrids. This would mean that the ultimate phenotype of tumor hybrids is not purely random, but could be driven to a certain phenotype dependent on the specific spatial tumor microenvironment.
Intratumoral heterogeneity is a hallmark of cancer [95] and the main reason for acquired resistance during therapy, which enables some tumor cells to survive treatment and facilitates the development of new therapy-resistant phenotypes [90]. Given that cell–cell fusion together with mutations is a potent driver of diversification, clonal richness and an overall enhanced tumoral heterogeneity [36], it can be concluded that the process of cell-cell fusion is involved in the origin of therapy-resistant phenotypes, which is in line with the hypothesis that cell–cell fusion could give rise to tumor hybrids exhibiting novel properties, such as an enhanced drug resistance [96][97][98][99][100]. Hence, the process of cell–cell fusion might be a suitable target for novel anti-cancer therapies. The inhibition of homotypic and heterotypic cell–cell fusion events should be correlated with a decreased diversification and clonal richness, and an overall reduced intratumoral heterogeneity.
While this conclusion sounds conclusive, the reality is much more complex. To understand the process of cell–cell fusion better and its impact on tumor progression, it is mandatory to clarify how many tumor hybrids will reside in the primary tumor, in the circulation and in metastases. This, however, requires appropriate valid fusion or hybrid cell markers. Cell-cell fusion could give rise to binucleated cells, but also cytokinesis errors, endoreduplication or entosis [101][102]. The expression of hematopoietic lineage markers could be related to a former cell-cell fusion event [37][52][103][104][105], but to genomic instability as well. Thus far, tumor hybrids have been only clearly identified by genetic markers in cancer patients with a former BMT history [37][106][66][67][107], but these cases are pretty rare. Mathematical data suggesting that even a low fusion probability could have a pronounced effect on intratumoral heterogeneity [36] are promising, but need to be validated.
Likewise, cell-cell fusion is still a not well-understood process, which applies to both proteins and conditions that regulate and induce the merging of two (and more) cells. Do cancer cells fuse in a uniform manner through a conserved mechanism or does the fusion machinery differ between different types of cancer? Without this knowledge, the development of appropriate therapies would not be possible. Moreover, the correlation between cell–cell fusion, an enhanced intratumoral heterogeneity and therapy resistance suggests that therapy-resistant cancer cells predominantly originate from hybridization events. However, therapy-resistant cancer cells could also originate due to genetic and epigenetic alterations or interactions with the microenvironment without fusion [90], which is another point that has be clarified before specific therapies could be developed.
References
- Aguilar, P.S.; Baylies, M.K.; Fleissner, A.; Helming, L.; Inoue, N.; Podbilewicz, B.; Wang, H.; Wong, M. Genetic basis of cell-cell fusion mechanisms. Trends Genet. 2013, 29, 427–437.
- Hernandez, J.M.; Podbilewicz, B. The hallmarks of cell-cell fusion. Development 2017, 144, 4481–4495.
- Zhou, X.; Platt, J.L. Molecular and cellular mechanisms of Mammalian cell fusion. Adv. Exp. Med. Biol. 2011, 713, 33–64.
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522.
- Brukman, N.G.; Uygur, B.; Podbilewicz, B.; Chernomordik, L.V. How cells fuse. J. Cell Biol. 2019, 218, 1436–1451.
- Dornen, J.; Sieler, M.; Weiler, J.; Keil, S.; Dittmar, T. Cell Fusion-Mediated Tissue Regeneration as an Inducer of Polyploidy and Aneuploidy. Int. J. Mol. Sci. 2020, 21, 1811.
- Huppertz, B.; Bartz, C.; Kokozidou, M. Trophoblast fusion: Fusogenic proteins, syncytins and ADAMs, and other prerequisites for syncytial fusion. Micron 2006, 37, 509–517.
- Mi, S.; Lee, X.; Li, X.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789.
- Muir, A.; Lever, A.M.; Moffett, A. Human endogenous retrovirus-W envelope (syncytin) is expressed in both villous and extravillous trophoblast populations. J. Gen. Virol. 2006, 87, 2067–2071.
- Millay, D.P.; O’Rourke, J.R.; Sutherland, L.B.; Bezprozvannaya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 2013, 499, 301–305.
- Leikina, E.; Gamage, D.G.; Prasad, V.; Goykhberg, J.; Crowe, M.; Diao, J.; Kozlov, M.M.; Chernomordik, L.V.; Millay, D.P. Myomaker and Myomerger Work Independently to Control Distinct Steps of Membrane Remodeling during Myoblast Fusion. Dev. Cell 2018, 46, 767–780.e767.
- Kato, K.; Satouh, Y.; Nishimasu, H.; Kurabayashi, A.; Morita, J.; Fujihara, Y.; Oji, A.; Ishitani, R.; Ikawa, M.; Nureki, O. Structural and functional insights into IZUMO1 recognition by JUNO in mammalian fertilization. Nat. Commun. 2016, 7, 12198.
- Inoue, N.; Ikawa, M.; Isotani, A.; Okabe, M. The immunoglobulin superfamily protein Izumo is required for sperm to fuse with eggs. Nature 2005, 434, 234–238.
- Bianchi, E.; Wright, G.J. Cross-species fertilization: The hamster egg receptor, Juno, binds the human sperm ligand, Izumo1. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140101.
- Bjerregaard, B.; Holck, S.; Christensen, I.J.; Larsson, L.I. Syncytin is involved in breast cancer-endothelial cell fusions. Cell. Mol. Life Sci. 2006, 63, 1906–1911.
- Strick, R.; Ackermann, S.; Langbein, M.; Swiatek, J.; Schubert, S.W.; Hashemolhosseini, S.; Koscheck, T.; Fasching, P.A.; Schild, R.L.; Beckmann, M.W.; et al. Proliferation and cell-cell fusion of endometrial carcinoma are induced by the human endogenous retroviral Syncytin-1 and regulated by TGF-beta. J. Mol. Med. 2007, 85, 23–38.
- Yan, T.L.; Wang, M.; Xu, Z.; Huang, C.M.; Zhou, X.C.; Jiang, E.H.; Zhao, X.P.; Song, Y.; Song, K.; Shao, Z.; et al. Up-regulation of syncytin-1 contributes to TNF-alpha-enhanced fusion between OSCC and HUVECs partly via Wnt/beta-catenin-dependent pathway. Sci. Rep. 2017, 7, 40983.
- Yu, H.; Liu, T.; Zhao, Z.; Chen, Y.; Zeng, J.; Liu, S.; Zhu, F. Mutations in 3′-long terminal repeat of HERV-W family in chromosome 7 upregulate syncytin-1 expression in urothelial cell carcinoma of the bladder through interacting with c-Myb. Oncogene 2014, 33, 3947–3958.
- Larsson, L.I.; Holck, S.; Christensen, I.J. Prognostic role of syncytin expression in breast cancer. Hum. Pathol. 2007, 38, 726–731.
- Melzer, C.; von der Ohe, J.; Hass, R. In vitro fusion of normal and neoplastic breast epithelial cells with human mesenchymal stroma/stem cells (MSC) partially involves TNF receptor signaling. Stem Cells 2018, 36, 977–989.
- Davies, P.S.; Powell, A.E.; Swain, J.R.; Wong, M.H. Inflammation and proliferation act together to mediate intestinal cell fusion. PLoS ONE 2009, 4, e6530.
- Johansson, C.B.; Youssef, S.; Koleckar, K.; Holbrook, C.; Doyonnas, R.; Corbel, S.Y.; Steinman, L.; Rossi, F.M.; Blau, H.M. Extensive fusion of haematopoietic cells with Purkinje neurons in response to chronic inflammation. Nat. Cell Biol. 2008, 10, 575–583.
- McNally, A.K.; Anderson, J.M. Macrophage fusion and multinucleated giant cells of inflammation. Adv. Exp. Med. Biol. 2011, 713, 97–111.
- Weiler, J.; Dittmar, T. Minocycline impairs TNF-alpha-induced cell fusion of M13SV1-Cre cells with MDA-MB-435-pFDR1 cells by suppressing NF-kappaB transcriptional activity and its induction of target-gene expression of fusion-relevant factors. Cell Commun. Signal. 2019, 17, 71.
- Weiler, J.; Mohr, M.; Zanker, K.S.; Dittmar, T. Matrix metalloproteinase-9 (MMP9) is involved in the TNF-alpha-induced fusion of human M13SV1-Cre breast epithelial cells and human MDA-MB-435-pFDR1 cancer cells. Cell Commun. Signal. 2018, 16, 14.
- Skokos, E.A.; Charokopos, A.; Khan, K.; Wanjala, J.; Kyriakides, T.R. Lack of TNF-alpha-induced MMP-9 production and abnormal E-cadherin redistribution associated with compromised fusion in MCP-1-null macrophages. Am. J. Pathol. 2011, 178, 2311–2321.
- Hotokezaka, H.; Sakai, E.; Ohara, N.; Hotokezaka, Y.; Gonzales, C.; Matsuo, K.; Fujimura, Y.; Yoshida, N.; Nakayama, K. Molecular analysis of RANKL-independent cell fusion of osteoclast-like cells induced by TNF-alpha, lipopolysaccharide, or peptidoglycan. J. Cell. Biochem. 2007, 101, 122–134.
- Takahashi, Y.; Bigler, D.; Ito, Y.; White, J.M. Sequence-specific interaction between the disintegrin domain of mouse ADAM 3 and murine eggs: Role of beta1 integrin-associated proteins CD9, CD81, and CD98. Mol. Biol. Cell 2001, 12, 809–820.
- Aghababaei, M.; Hogg, K.; Perdu, S.; Robinson, W.P.; Beristain, A.G. ADAM12-directed ectodomain shedding of E-cadherin potentiates trophoblast fusion. Cell Death Differ. 2015, 22, 1970–1984.
- Rodriguez, D.; Morrison, C.J.; Overall, C.M. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 2010, 1803, 39–54.
- Willkomm, L.; Bloch, W. State of the art in cell-cell fusion. Methods Mol. Biol. 2015, 1313, 1–19.
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545.
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867.
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444.
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659.
- Miroshnychenko, D.; Baratchart, E.; Ferrall-Fairbanks, M.C.; Velde, R.V.; Laurie, M.A.; Bui, M.M.; Tan, A.C.; Altrock, P.M.; Basanta, D.; Marusyk, A. Spontaneous cell fusions as a mechanism of parasexual recombination in tumour cell populations. Nat. Ecol. Evol. 2021, 5, 379–391.
- Gast, C.E.; Silk, A.D.; Zarour, L.; Riegler, L.; Burkhart, J.G.; Gustafson, K.T.; Parappilly, M.S.; Roh-Johnson, M.; Goodman, J.R.; Olson, B.; et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 2018, 4, eaat7828.
- Melzer, C.; von der Ohe, J.; Hass, R. In Vivo Cell Fusion between Mesenchymal Stroma/Stem-Like Cells and Breast Cancer Cells. Cancers 2019, 11, 185.
- Fortuna, M.B.; Dewey, M.J.; Furmanski, P. Cell fusion in tumor development and progression: Occurrence of cell fusion in primary methylcholanthrene-induced tumorigenesis. Int. J. Cancer 1989, 44, 731–737.
- Yan, B.; Wang, J.; Liu, L. Chemotherapy promotes tumour cell hybridization in vivo. Tumour Biol. 2015, 37, 5025–5030.
- Weiler, J.; Dittmar, T. Cell Fusion in Human Cancer: The Dark Matter Hypothesis. Cells 2019, 8, 132.
- Wakeling, W.F.; Greetham, J.; Bennett, D.C. Efficient spontaneous fusion between some co-cultured cells, especially murine melanoma cells. Cell Biol. Int. 1994, 18, 207–210.
- Powell, A.E.; Anderson, E.C.; Davies, P.S.; Silk, A.D.; Pelz, C.; Impey, S.; Wong, M.H. Fusion between Intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011, 71, 1497–1505.
- Rachkovsky, M.; Sodi, S.; Chakraborty, A.; Avissar, Y.; Bolognia, J.; McNiff, J.M.; Platt, J.; Bermudes, D.; Pawelek, J. Melanoma x macrophage hybrids with enhanced metastatic potential. Clin. Exp. Metastasis 1998, 16, 299–312.
- Jacobsen, B.M.; Harrell, J.C.; Jedlicka, P.; Borges, V.F.; Varella-Garcia, M.; Horwitz, K.B. Spontaneous fusion with, and transformation of mouse stroma by, malignant human breast cancer epithelium. Cancer Res. 2006, 66, 8274–8279.
- Chakraborty, A.K.; Sodi, S.; Rachkovsky, M.; Kolesnikova, N.; Platt, J.T.; Bolognia, J.L.; Pawelek, J.M. A spontaneous murine melanoma lung metastasis comprised of host x tumor hybrids. Cancer Res. 2000, 60, 2512–2519.
- Su, Y.; Subedee, A.; Bloushtain-Qimron, N.; Savova, V.; Krzystanek, M.; Li, L.; Marusyk, A.; Tabassum, D.P.; Zak, A.; Flacker, M.J.; et al. Somatic Cell Fusions Reveal Extensive Heterogeneity in Basal-like Breast Cancer. Cell Rep. 2015, 11, 1549–1563.
- Lu, X.; Kang, Y. Efficient acquisition of dual metastasis organotropism to bone and lung through stable spontaneous fusion between MDA-MB-231 variants. Proc. Natl. Acad. Sci. USA 2009, 106, 9385–9390.
- Clawson, G.A.; Matters, G.L.; Xin, P.; Imamura-Kawasawa, Y.; Du, Z.; Thiboutot, D.M.; Helm, K.F.; Neves, R.I.; Abraham, T. Macrophage-Tumor Cell Fusions from Peripheral Blood of Melanoma Patients. PLoS ONE 2015, 10, e0134320.
- Dittmar, T.; Schwitalla, S.; Seidel, J.; Haverkampf, S.; Reith, G.; Meyer-Staeckling, S.; Brandt, B.H.; Niggemann, B.; Zanker, K.S. Characterization of hybrid cells derived from spontaneous fusion events between breast epithelial cells exhibiting stem-like characteristics and breast cancer cells. Clin. Exp. Metastasis 2011, 28, 75–90.
- Hass, R.; von der Ohe, J.; Ungefroren, H. Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation. Cancers 2019, 11, 1432.
- Ramakrishnan, M.; Mathur, S.R.; Mukhopadhyay, A. Fusion derived epithelial cancer cells express hematopoietic markers and contribute to stem cell and migratory phenotype in ovarian carcinoma. Cancer Res. 2013, 73, 5360–5370.
- Shabo, I.; Midtbo, K.; Andersson, H.; Akerlund, E.; Olsson, H.; Wegman, P.; Gunnarsson, C.; Lindstrom, A. Macrophage traits in cancer cells are induced by macrophage-cancer cell fusion and cannot be explained by cellular interaction. BMC Cancer 2015, 15, 922.
- Zhang, L.N.; Kong, C.F.; Zhao, D.; Cong, X.L.; Wang, S.S.; Ma, L.; Huang, Y.H. Fusion with mesenchymal stem cells differentially affects tumorigenic and metastatic abilities of lung cancer cells. J. Cell. Physiol. 2018, 234, 3570–3582.
- Zhang, L.N.; Zhang, D.D.; Yang, L.; Gu, Y.X.; Zuo, Q.P.; Wang, H.Y.; Xu, J.; Liu, D.X. Roles of cell fusion between mesenchymal stromal/stem cells and malignant cells in tumor growth and metastasis. FEBS J. 2020, 288, 1447–1456.
- Kemeny, L.V.; Kurgyis, Z.; Buknicz, T.; Groma, G.; Jakab, A.; Zanker, K.; Dittmar, T.; Kemeny, L.; Nemeth, I.B. Melanoma Cells Can Adopt the Phenotype of Stromal Fibroblasts and Macrophages by Spontaneous Cell Fusion in Vitro. Int. J. Mol. Sci. 2016, 17, 826.
- Melzer, C.; Ohe, J.V.; Hass, R. Altered Tumor Plasticity after Different Cancer Cell Fusions with MSC. Int. J. Mol. Sci. 2020, 21, 8347.
- Lindstrom, A.; Midtbo, K.; Arnesson, L.G.; Garvin, S.; Shabo, I. Fusion between M2-macrophages and cancer cells results in a subpopulation of radioresistant cells with enhanced DNA-repair capacity. Oncotarget 2017, 8, 51370–51386.
- Lizier, M.; Anselmo, A.; Mantero, S.; Ficara, F.; Paulis, M.; Vezzoni, P.; Lucchini, F.; Pacchiana, G. Fusion between cancer cells and macrophages occurs in a murine model of spontaneous neu+ breast cancer without increasing its metastatic potential. Oncotarget 2016, 7, 60793–60806.
- Noubissi, F.K.; Harkness, T.; Alexander, C.M.; Ogle, B.M. Apoptosis-induced cancer cell fusion: A mechanism of breast cancer metastasis. FASEB J. 2015, 29, 4036–4045.
- Zeng, C.; Zhang, Y.; Park, S.C.; Eun, J.R.; Nguyen, N.T.; Tschudy-Seney, B.; Jung, Y.J.; Theise, N.D.; Zern, M.A.; Duan, Y. CD34 Liver Cancer Stem Cells Were Formed by Fusion of Hepatobiliary Stem/Progenitor Cells with Hematopoietic Precursor-Derived Myeloid Intermediates. Stem Cells Dev. 2015, 24, 2467–2478.
- Luo, F.; Liu, T.; Wang, J.; Li, J.; Ma, P.; Ding, H.; Feng, G.; Lin, D.; Xu, Y.; Yang, K. Bone marrow mesenchymal stem cells participate in prostate carcinogenesis and promote growth of prostate cancer by cell fusion in vivo. Oncotarget 2016, 7, 30924–30934.
- Goldenberg, D.M.; Pavia, R.A.; Tsao, M.C. In vivo hybridisation of human tumour and normal hamster cells. Nature 1974, 250, 649–651.
- Sottile, F.; Aulicino, F.; Theka, I.; Cosma, M.P. Mesenchymal stem cells generate distinct functional hybrids in vitro via cell fusion or entosis. Sci. Rep. 2016, 6, 36863.
- Dornen, J.; Myklebost, O.; Dittmar, T. Cell Fusion of Mesenchymal Stem/Stromal Cells and Breast Cancer Cells Leads to the Formation of Hybrid Cells Exhibiting Diverse and Individual (Stem Cell) Characteristics. Int. J. Mol. Sci. 2020, 21, 9636.
- LaBerge, G.S.; Duvall, E.; Grasmick, Z.; Haedicke, K.; Pawelek, J. A Melanoma Lymph Node Metastasis with a Donor-Patient Hybrid Genome following Bone Marrow Transplantation: A Second Case of Leucocyte-Tumor Cell Hybridization in Cancer Metastasis. PLoS ONE 2017, 12, e0168581.
- Lazova, R.; Laberge, G.S.; Duvall, E.; Spoelstra, N.; Klump, V.; Sznol, M.; Cooper, D.; Spritz, R.A.; Chang, J.T.; Pawelek, J.M. A Melanoma Brain Metastasis with a Donor-Patient Hybrid Genome following Bone Marrow Transplantation: First Evidence for Fusion in Human Cancer. PLoS ONE 2013, 8, e66731.
- Bjerkvig, R.; Tysnes, B.B.; Aboody, K.S.; Najbauer, J.; Terzis, A.J. Opinion: The origin of the cancer stem cell: Current controversies and new insights. Nat. Rev. Cancer 2005, 5, 899–904.
- Duncan, A.W.; Hickey, R.D.; Paulk, N.K.; Culberson, A.J.; Olson, S.B.; Finegold, M.J.; Grompe, M. Ploidy reductions in murine fusion-derived hepatocytes. PLoS Genet. 2009, 5, e1000385.
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Hanlon Newell, A.E.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010, 467, 707–710.
- Hass, R.; von der Ohe, J.; Ungefroren, H. Impact of the Tumor Microenvironment on Tumor Heterogeneity and Consequences for Cancer Cell Plasticity and Stemness. Cancers 2020, 12, 3716.
- Li, R.; Sonik, A.; Stindl, R.; Rasnick, D.; Duesberg, P. Aneuploidy versus gene mutation hypothesis of cancer: Recent study claims mutation, but is found to support aneuploidy. Proc. Natl. Acad. Sci. USA 2000, 97, 3236–3241.
- Sun, W.; Kang, K.S.; Morita, I.; Trosko, J.E.; Chang, C.C. High susceptibility of a human breast epithelial cell type with stem cell characteristics to telomerase activation and immortalization. Cancer Res. 1999, 59, 6118–6123.
- Ly, P.; Cleveland, D.W. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017, 27, 917–930.
- Rode, A.; Maass, K.K.; Willmund, K.V.; Lichter, P.; Ernst, A. Chromothripsis in cancer cells: An update. Int. J. Cancer 2016, 138, 2322–2333.
- Chunduri, N.K.; Storchova, Z. The diverse consequences of aneuploidy. Nat. Cell Biol. 2019, 21, 54–62.
- Zhou, X.; Merchak, K.; Lee, W.; Grande, J.P.; Cascalho, M.; Platt, J.L. Cell Fusion Connects Oncogenesis with Tumor Evolution. Am. J. Pathol. 2015, 185, 2049–2060.
- Dittmar, T.; Nagler, C.; Schwitalla, S.; Reith, G.; Niggemann, B.; Zanker, K.S. Recurrence cancer stem cells-made by cell fusion? Med. Hypotheses 2009, 73, 542–547.
- Fahlbusch, S.S.; Keil, S.; Epplen, J.T.; Zanker, K.S.; Dittmar, T. Comparison of hybrid clones derived from human breast epithelial cells and three different cancer cell lines regarding in vitro cancer stem/initiating cell properties. BMC Cancer 2020, 20, 446.
- Delespaul, L.; Merle, C.; Lesluyes, T.; Lagarde, P.; Le Guellec, S.; Perot, G.; Baud, J.; Carlotti, M.; Danet, C.; Fevre, M.; et al. Fusion-mediated chromosomal instability promotes aneuploidy patterns that resemble human tumors. Oncogene 2019, 38, 6083–6094.
- Delespaul, L.; Gelabert, C.; Lesluyes, T.; Le Guellec, S.; Perot, G.; Leroy, L.; Baud, J.; Merle, C.; Lartigue, L.; Chibon, F. Cell-cell fusion of mesenchymal cells with distinct differentiations triggers genomic and transcriptomic remodelling toward tumour aggressiveness. Sci. Rep. 2020, 10, 21634.
- Lartigue, L.; Merle, C.; Lagarde, P.; Delespaul, L.; Lesluyes, T.; Le Guellec, S.; Perot, G.; Leroy, L.; Coindre, J.M.; Chibon, F. Genome remodeling upon mesenchymal tumor cell fusion contributes to tumor progression and metastatic spread. Oncogene 2020, 39, 4198–4211.
- Miller, F.R.; McInerney, D.; Rogers, C.; Miller, B.E. Spontaneous fusion between metastatic mammary tumor subpopulations. J. Cell. Biochem. 1988, 36, 129–136.
- Wang, R.; Sun, X.; Wang, C.Y.; Hu, P.; Chu, C.Y.; Liu, S.; Zhau, H.E.; Chung, L.W. Spontaneous cancer-stromal cell fusion as a mechanism of prostate cancer androgen-independent progression. PLoS ONE 2012, 7, e42653.
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009.
- Jiang, E.; Yan, T.; Xu, Z.; Shang, Z. Tumor Microenvironment and Cell Fusion. Biomed Res. Int. 2019, 2019, 5013592.
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437.
- Hoefflin, R.; Lahrmann, B.; Warsow, G.; Hubschmann, D.; Spath, C.; Walter, B.; Chen, X.; Hofer, L.; Macher-Goeppinger, S.; Tolstov, Y.; et al. Spatial niche formation but not malignant progression is a driving force for intratumoural heterogeneity. Nat. Commun. 2016, 7, 1–12.
- Lloyd, M.C.; Cunningham, J.J.; Bui, M.M.; Gillies, R.J.; Brown, J.S.; Gatenby, R.A. Darwinian Dynamics of Intratumoral Heterogeneity: Not Solely Random Mutations but Also Variable Environmental Selection Forces. Cancer Res. 2016, 76, 3136–3144.
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484.
- Li, L.; Neaves, W.B. Normal stem cells and cancer stem cells: The niche matters. Cancer Res. 2006, 66, 4553–4557.
- He, X.; Li, B.; Shao, Y.; Zhao, N.; Hsu, Y.; Zhang, Z.; Zhu, L. Cell fusion between gastric epithelial cells and mesenchymal stem cells results in epithelial-to-mesenchymal transition and malignant transformation. BMC Cancer 2015, 15, 24.
- Gauck, D.; Keil, S.; Niggemann, B.; Zanker, K.S.; Dittmar, T. Hybrid clone cells derived from human breast epithelial cells and human breast cancer cells exhibit properties of cancer stem/initiating cells. BMC Cancer 2017, 17, 515.
- Nagler, C.; Hardt, C.; Zänker, K.S.; Dittmar, T. Co-cultivation of murine BMDCs with 67NR mouse mammary carcinoma cells give rise to highly drug resistant hybrid cells. Cancer Cell Int. 2011, 11, 21.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Duelli, D.; Lazebnik, Y. Cell fusion: A hidden enemy? Cancer Cell 2003, 3, 445–448.
- Carloni, V.; Mazzocca, A.; Mello, T.; Galli, A.; Capaccioli, S. Cell fusion promotes chemoresistance in metastatic colon carcinoma. Oncogene 2013, 32, 2649–2660.
- Mirzayans, R.; Murray, D. Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence. Int. J. Mol. Sci. 2020, 21, 1308.
- Mohr, M.; Zaenker, K.S.; Dittmar, T. Fusion in cancer: An explanatory model for aneuploidy, metastasis formation, and drug resistance. Methods Mol. Biol. 2015, 1313, 21–40.
- Uygur, B.; Leikina, E.; Melikov, K.; Villasmil, R.; Verma, S.K.; Vary, C.P.H.; Chernomordik, L.V. Interactions with Muscle Cells Boost Fusion, Stemness, and Drug Resistance of Prostate Cancer Cells. Mol. Cancer Res. 2019, 17, 806–820.
- Fais, S.; Overholtzer, M. Cell-in-cell phenomena in cancer. Nat. Rev. Cancer 2018, 18, 758–766.
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45.
- Shabo, I.; Stal, O.; Olsson, H.; Dore, S.; Svanvik, J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int. J. Cancer 2008, 123, 780–786.
- Shabo, I.; Olsson, H.; Sun, X.F.; Svanvik, J. Expression of the macrophage antigen CD163 in rectal cancer cells is associated with early local recurrence and reduced survival time. Int. J. Cancer 2009, 125, 1826–1831.
- Shabo, I.; Olsson, H.; Stal, O.; Svanvik, J. Breast cancer expression of DAP12 is associated with skeletal and liver metastases and poor survival. Clin. Breast Cancer 2013, 13, 371–377.
- Chakraborty, A.; Lazova, R.; Davies, S.; Backvall, H.; Ponten, F.; Brash, D.; Pawelek, J. Donor DNA in a renal cell carcinoma metastasis from a bone marrow transplant recipient. Bone Marrow Transplant. 2004, 34, 183–186.
- Yilmaz, Y.; Lazova, R.; Qumsiyeh, M.; Cooper, D.; Pawelek, J. Donor Y chromosome in renal carcinoma cells of a female BMT recipient: Visualization of putative BMT-tumor hybrids by FISH. Bone Marrow Transplant. 2005, 35, 1021–1024.



