 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anna Pieniążek | + 2082 word(s) | 2082 | 2021-06-17 09:38:00 | | | |
| 2 | Camila Xu | + 22 word(s) | 2104 | 2021-06-28 05:23:27 | | |
Video Upload Options
Some uremic toxins such as indol acetic acid, indoxyl sulfate, p-cresyl sulfate hippuric acid, phenyl sulfate, trimethylamine N-oxide, and hydrogen sulfide in the blood of CKD patients come from the metabolism of bacteria in the gut. As a result of the conducted research, the list of potential uremic toxins is constantly being modified. For example, it is suggested that the current list of cytokines that are considered potential uremic toxins is incomplete and excessive.
1. Introduction
Chronic kidney disease leads to uremia associated with the accumulation of uremic toxins in the body. That causes unfavorable changes in the cardiovascular system, some of them lead to chronic inflammation, endothelial dysfunction, damage to the mitochondria and, consequently, oxidative stress. In 2003, the European Uremic Toxin Work Group (EUTox) classified 90 substances as uremic toxins [1]. Among them, there are advanced glycation end products (AGE), phenolic derivatives (including p-cresol), indole derivatives, hippurates, polyamines, peptides, homocysteine, 3-carboxy-4-methyl-5-propyl-2-furanopropionic acid (CMPF), and trimethylamine-N-oxide (TMAO).
The toxins dissolved in uremia were classified based on their size and binding properties. Free, water-soluble compounds of low molecular weight (500 g/mol) accounted for 46% of the 88 solutes; 28% of the solutes were particles with a mass of 500–60,000 g/mol, and 24% of the solutes were associated with proteins solutes [2].
Most of them are peptides that are difficult to remove during hemodialysis unless the pore size of the dialyzer is large enough. In turn, removal of toxins associated with proteins from the body of a patient with CKD is almost impossible using dialysis [3].
Some uremic toxins such as indol acetic acid, indoxyl sulfate, p-cresyl sulfate hippuric acid, phenyl sulfate, trimethylamine N-oxide, and hydrogen sulfide in the blood of CKD patients come from the metabolism of bacteria in the gut [4][5]. As a result of the conducted research, the list of potential uremic toxins is constantly being modified. For example, it is suggested that the current list of cytokines that are considered potential uremic toxins is incomplete and excessive [6].
Many toxins presented in this review can release ROS enhancing oxidative stress in organism of CKD patients. Uremic toxins and oxidative stress lead to modifications of proteins, lipids, and DNA. Some of these modifications are protein carbonyl compounds as products of protein oxidation, lipids oxidation (thiobarbituric acid reactive substances (TBARS), In addition, hemodialysis worsens these processes, especially those related to oxidative stress [7].
In this review, we summarize recent research showing that uremic toxins have a biological effect on the cells and tissues of CKD patients as well as on the development of the disease. Modified biomolecules as a result of carbamylation, oxidative stress, and covalent adducts formation of uremic toxins with biopolymers can lead to cardiovascular diseases, increased risk, and mortality in patients with CKD [8][9][10].
2. Urea Toxicity
It is known that elevated urea levels can lead to molecular changes related to insulin resistance, generation of reactive oxygen species, apoptosis, and disruption of the intestinal protective barrier [11]. Urea is one of the earliest known uremic toxins in chronic kidney disease (CKD); its concentration in patients is about 5–7 times higher than in healthy individuals. Isocyanic acid reacts with the thiol and amino groups of proteins, amino acids, sugars, etc. Modifications also apply to amino acids such as cysteine, histidine, arginine, lysine, valine, tryptophan, and threonine, as well as proteins containing these amino acids [12][13].
Carbamylation also affects such macromolecules as hemoglobin, plasma lipoproteins, albumin, membrane proteins, and erythropoietin in patients with chronic renal failure [14]. For example, carbamylated hemoglobin concentration was about three times higher in people with chronic renal failure undergoing both hemodialysis or peritoneal dialysis patients [8]. In addition, changes in the structure of hemoglobin and non-hem proteins of erythrocyte were observed in patients with CKD [9]. In turn, carbamylated low density lipoproteins (LDL) affect atherosclerotic through the participation in the formation of foam cells, induction of apoptosis of endothelial cells, and proliferation of smooth muscle cells; myeloperoxidase is involved in this process [15][16][17].
The carbamylation process is thought to be responsible for the post-translational modifications of proteins that are related to atherogenesis and other functional changes. In clinical trials, carbamylated proteins, amino acids, and other compounds have been associated with cardiovascular disease and overall morbidity and mortality [11]. It was shown that treatment of 3T3-L1 adipocytes with urea at concentrations similar to those in CKD patients led to the production of ROS, caused insulin resistance, increased the expression of adipokines, retinol-binding protein 4 (RBP4), and resistin, and increased the level of O-linked N-acetylglucosamine (O-GlcNAc) In-vivo studies using uremic mice, a mouse model of surgically induced renal failure, showed ROS production, modification of insulin signaling molecules by O-GlcNAc, and an increased expression of RBP4 and resistin in visceral adipose tissue.
In vitro, after treatment of red blood cells (RBC) with urea, there was an increase in the fluidity of the lipid membrane in the polar and hydrophobic regions of the cores. The changes in the fluidity of membranes were accompanied by changes in the RBC membrane cytoskeleton. It seems that carbamylated proteins are more sensitive to oxidation than native proteins [18]. Carbamylated proteins may activate mesangial cells to a profibrogenic form, which may contribute to the development of renal failure [19].
One of the products of protein carbamylation is homocitrulline, which is formed by binding isocyanic acid to the ε-NH2group of the side chain of lysine residues [10]. Nowadays, homocitrulline is an important biomarker in patients with uremia and its increase in concentration allows distinguishing acute from chronic renal failure and is a risk factor of mortality [20]. The observations presented here show that urea concentration in CKD patients, long considered insignificant, may lead to increased ROS production, cause insulin resistance in vitro and in vivo, and lead to cardiovascular disease and mortality in CKD patients.
This likely has to do with defective insulin secretion, which may be the result of a direct action of urea on pancreatic β cells. These results are indicative of an impaired insulin secretion in CKD caused by elevated blood urea levels that lead to an increase in O-GlcNCylation of islet protein and impair glycolysis [21]. A good way to reduce urea levels in CKD patients is nutritional therapy with low protein content. It has been demonstrated that low protein diet may decrease urea levels and reduce protein carbamylation as well as cyanate production [12].
3. Polyamines
The research carried out in 1983 showed that in patients with CKD, the level of polyamines (putrescine, spermine, and spermidine) (Figure 1) increases significantly in comparison to healthy volunteers [22].
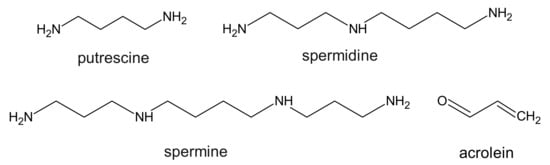
17–39 µg/L in healthy people and 50–84 µg/L in patients with CKD [23][24][25], which means that in CKD patients, the level of free acrolein was 3–5-fold higher when compared to control subjects. The authors also suggest that oxidative stress in patients with CKD may contribute to an increase in acrolein production during hemodialysis, and thus reduce the kinetics of its removal [24]. On the other hand, a recent report demonstrated that in non-hemodialyzed patients, the levels of acrolein and protein-conjugated acrolein were even higher than in hemodialyzed patients [26]. Considering the fact that polyamines are necessary for cellular growth, some research suggests that it is in fact acrolein and not polyamines, that functions as a uremic toxin in patients with CKD.
In high doses of acrolein, the concentration of GSH is low to provide cellular protection against this toxin [27]. Its reaction with the cysteine thiol group was about 110–150 times faster than HNE, a famous product of lipid peroxidation. Given its high affinity for thiols, it could be possible to lower its concentration by administering N-acetlcysteine or other thiols used in the treatment of heavy metal poisoning. Acrolein toxicity is related to the formation of covalent adducts of DNA and proteins via cysteinyl, histidyl, and lysyl residues as well as free N-terminal amino groups [28][29].
4. Implications of Uremic Toxins and Oxidative Stress in Cardiovascular Disease
In recent years, ROS that initiate oxidative stress have been shown to perform a key role in the development and maintenance of inflammation, contributing to the pathophysiology of many debilitating diseases such as cardiovascular diseases (CVD), diabetes, cancer or neurodegenerative diseases [30]. ROS affect all stages of the inflammatory response, releasing molecules from damaged tissues that act as endogenous signals of danger. These signals are recorded by the innate immune receptors Toll (TLR) and NOD-like (NLR) and activate signaling pathways that initiate an adaptive cellular response to such signals [31].
CKD is accompanied by oxidative stress and chronic inflammation [32]. Inflammation is caused by a variety of factors, such as increased production and decreased clearance of inflammatory cytokines, oxidative stress, acidosis, chronic and recurrent infections, also associated with dialysis. Figure 2 presents the complexity of the interactions between uremic toxins and oxidative stress in patients with CKD.
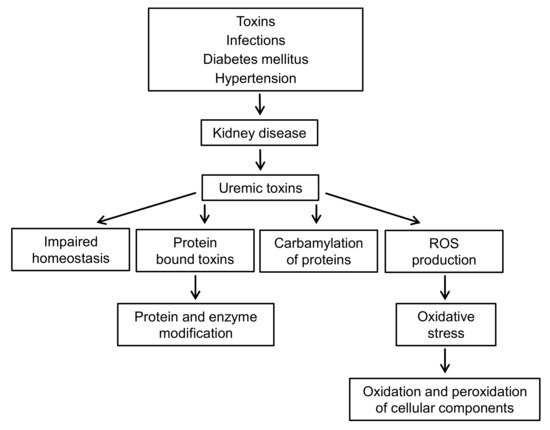
This applies to both the enzymatic defense related to the activity of enzymes such as superoxide dismutase (SOD1), catalase (Cat), glutathione peroxidase (GPx), and low molecular weight antioxidants, e.g., glutathione, ascorbic acid, α-tocopherol, β-carotene, ubiquinol, bilirubin, and others. In CKD, an impaired antioxidant system is observed, which is associated with decreased activity of GPx and Cat in the plasma, and increased activity of GPx and SOD1 (CuZnSOD) in RBC [33][34]. In other studies, decreased glutathione peroxidase activity and decreased SOD1 activity were observed [35]. Another marker of antioxidant activity was paraoxonase, which is an esterase enzyme related to high density lipoproteins (HDL) and works to protect LDL and HDL from oxidation.
Moreover, in CKD, there is a high concentration of uric acid in the plasma (hyperuricemia). Uric acid synthesis can promote oxidative stress directly through the activity of xanthine oxidoreductase [36]. This enzyme is synthesized as xanthine dehydrogenase and is converted to xanthine oxidase [37]. The low biocompatibility of dialysis membranes leads to the activation of neutrophils and a respiratory burst in which superoxide anion radical, hydrogen peroxide, hydroxyl radicals, singlet oxygen, nitric oxide, and hypochlorous acid are produced [38].
Impaired glomerular filtration of the kidneys leads to the retention of toxic solutes that affect all organs of the body. The consequences of kidney dysfunction are CVD and infections, which increase the morbidity and mortality of patients with CKD. The roles of cells such as polymorphonuclear leukocytes, monocytes/macrophages, lymphocytes, and antigen presenting cells in maintaining an effective immune response are disrupted. As a result, their normal response may be impaired, leading to infectious diseases or CVD [39].
Uremic toxins, oxidative stress, and permanent inflammation lead to a CVD in patients with CKD [40]. Both CVD and CKD, the so-called ”cardiorenal or renocardiac syndrome”, are directly associated with increased cardiovascular morbidity and mortality [41]. About half of all deaths of CKD patients are thought to be a direct consequence of CVD. Successful kidney transplantation significantly reduces the risk of death compared to long-term dialysis treatment, but also in this group of patients, the incidence of CVD remains high and amounts to approximately 20–35% of mortality in patients with kidney transplants [42].
CKD patients have cardiac arrhythmias, especially atrial fibrillation. (decreased left ventricular contraction/expansion function leading to heart failure) Initially, this condition is associated with left ventricular hypertrophy (LVH) and the characteristic fibrosis of the heart muscle, thickening of the wall of arterioles inside the heart muscle, and diastolic dysfunction, followed by systolic dysfunction, leading to overt heart failure at a later stage An additional risk factor for vascular diseases in CKD is hyperuricemia, which is associated with coronary artery disease, left ventricular hypertrophy, atrial fibrillation, myocardial infarction, and ischemic stroke [43][44][45].
In patients with CKD, accelerated atherosclerosis and reduced bioavailability of nitric oxide in vascular tissues are observed. The overproduction of ROS in CKD promotes reduced bioavailability of nitric oxide, which quickly reacts with the superoxide anion to form peroxynitrite, which in turn leads to oxidative stress and endothelial cell dysfunction, and consequently to atherosclerosis. Moreover, in CKD, the balance between pro and anti-inflammatory factors and between pro and anti-apoptotic factors is disturbed [39]. It has been shown that LDL isolated from CKD patients is more susceptible to peroxidation in vitro than LDL from healthy subjects [46].
Moreover, patients with high levels of TMAO in plasma are at increased risk of myocardial infarction or stroke [47]. CKD patients have significantly higher rates of the diagnosis of sepsis compared to patients without diagnosed CKD. Bacterial infections are the leading cause of hospitalization of CKD patients and are related to vascular access, which is the most common source of bacteremia in hemodialyzed patients. Sepsis is associated with an increased risk of thrombosis, myocardial infarction, congestive heart failure, stroke, and peripheral arterial disease [48][49].
Recently, it has been shown that the mortality rate of COVID-19 is higher in the group of patients with chronic kidney disease. This also applies to the acute kidney injury from COVID-19 infection [50].
References
- Vanholder, R.; de Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; de Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943.
- Duranton, F.; Cohen, G.; de Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270.
- Van Gelder, M.K.; Middel, I.R.; Vernooij, R.W.M.; Bots, M.L.; Verhaar, M.C.; Masereeuw, R.; Grooteman, M.P.; Nubé, M.J.; van den Dorpel, M.A.; Blankestijn, P.J.; et al. Protein-Bound Uremic Toxins in Hemodialysis Patients Relate to Residual Kidney Function, Are Not Influenced by Convective Transport, and Do Not Relate to Outcome. Toxins 2020, 12, 234.
- Tang, W.H.W. Trimethylamine N-Oxide as a Novel Therapeutic Target in CKD. J. Am. Soc. Nephrol. 2016, 27, 8–10.
- Gryp, T.; de Paepe, K.; Vanholder, R.; Kerckhof, F.-M.; van Biesen, W.; van de Wiele, T.; Verbeke, F.; Speeckaert, M.; Joossens, M.; Couttenye, M.M.; et al. Gut microbiota generation of protein-bound uremic toxins and related metabolites is not altered at different stages of chronic kidney disease. Kidney Int. 2020, 97, 1230–1242.
- Castillo-Rodríguez, E.; Pizarro-Sánchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory Cytokines as Uremic Toxins: “Ni Son Todos Los Que Estan, Ni Estan Todos Los Que Son”. Toxins 2017, 9, 114.
- Pieniazek, A.; Brzeszczynska, J.; Kruszynska, I.; Gwozdzinski, K. Investigation of albumin properties in patients with chronic renal failure. Free Radic. Res. 2009, 43, 1008–1018.
- Kairaitis, L.K.; Yuill, E.; Harris, D.C. Determinants of haemoglobin carbamylation in haemodialysis and peritoneal dialysis patients. Nephrol. Dial. Transpl. 2000, 15, 1431–1437.
- Pieniazek, A.; Gwozdzinski, K. Changes in the conformational state of hemoglobin in hemodialysed patients with chronic renal failure. Oxid. Med. Cell. Longev. 2015, 2015, 783073.
- Jaisson, S.; Kazes, I.; Desmons, A.; Fadel, F.; Oudart, J.-B.; Santos-Weiss, I.C.R.D.; Millart, H.; Touré, F.; Rieu, P.; Gillery, P. Homocitrulline as marker of protein carbamylation in hemodialyzed patients. Clin. Chim. Acta 2016, 460, 5–10.
- Vanholder, R.; Gryp, T.; Glorieux, G. Urea and chronic kidney disease: The comeback of the century? (in uraemia research). Nephrol. Dial. Transpl. 2018, 33, 4–12.
- Di Iorio, B.R.; Marzocco, S.; Bellasi, A.; de Simone, E.; Dal Piaz, F.; Rocchetti, M.T.; Cosola, C.; Di Micco, L.; Gesualdo, L. Nutritional therapy reduces protein carbamylation through urea lowering in chronic kidney disease. Nephrol. Dial. Transpl. 2018, 33, 804–813.
- Jin, K. Effects of amino acids and albumin on erythropoietin carbamoylation. Clin. Exp. Nephrol. 2013, 17, 575–581.
- Pieniążek, A.; Gwoździński, K. Karbamylacja białek—mechanizm, przyczyny i skutki. Postepy Hig. Med. Dosw. 2016, 70, 514–521.
- Jaisson, S.; Pietrement, C.; Gillery, P. Carbamylation-derived products: Bioactive compounds and potential biomarkers in chronic renal failure and atherosclerosis. Clin. Chem. 2011, 57, 1499–1505.
- Sirpal, S. Myeloperoxidase-mediated lipoprotein carbamylation as a mechanistic pathway for atherosclerotic vascular disease. Clin. Sci. 2009, 116, 681–695.
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184.
- Pieniazek, A.; Gwozdzinski, K.; Czepas, J. EPR study of erythrocyte properties after in vitro treatment with urea and hydrogen peroxide. Int. J. Sci. Res. 2014, 3, 491–494.
- Shaykh, M.; Pegoraro, A.A.; Mo, W.; Arruda, J.; Dunea, G.; Singh, A.K. Carbamylated proteins activate glomerular mesangial cells and stimulate collagen deposition. J. Lab. Clin. Med. 1999, 133, 302–308.
- Desmons, A.; Jaisson, S.; Pietrement, C.; Rieu, P.; Wynckel, A.; Gillery, P. Homocitrulline: A new marker for differentiating acute from chronic renal failure. Clin. Chem. Lab. Med. 2016, 54, 73–79.
- Koppe, L.; Nyam, E.; Vivot, K.; Manning Fox, J.E.; Dai, X.-Q.; Nguyen, B.N.; Trudel, D.; Attané, C.; Moullé, V.S.; MacDonald, P.E.; et al. Urea impairs β cell glycolysis and insulin secretion in chronic kidney disease. J. Clin. Investig. 2016, 126, 3598–3612.
- Saito, A.; Takagi, T.; Chung, T.G.; Ohta, K. Serum levels of polyamines in patients with chronic renal failure. Kidney Int. Suppl. 1983, 16, S234–S237.
- Igarashi, K.; Ueda, S.; Yoshida, K.; Kashiwagi, K. Polyamines in renal failure. Amino Acids 2006, 31, 477–483.
- Gugliucci, A.; Lunceford, N.; Kinugasa, E.; Ogata, H.; Schulze, J.; Kimura, S. Acrolein inactivates paraoxonase 1: Changes in free acrolein levels after hemodialysis correlate with increases in paraoxonase 1 activity in chronic renal failure patients. Clin. Chim. Acta 2007, 384, 105–112.
- Sakata, K.; Kashiwagi, K.; Sharmin, S.; Ueda, S.; Igarashi, K. Acrolein produced from polyamines as one of the uraemic toxins. Biochem. Soc. Trans. 2003, 31, 371–374.
- Igarashi, K.; Uemura, T.; Kashiwagi, K. Assessing acrolein for determination of the severity of brain stroke, dementia, renal failure, and Sjögren’s syndrome. Amino Acids 2020, 52, 119–127.
- Sthijns, M.M.J.P.E.; Randall, M.J.; Bast, A.; Haenen, G.R.M.M. Adaptation to acrolein through upregulating the protection by glutathione in human bronchial epithelial cells: The materialization of the hormesis concept. Biochem. Biophys. Res. Commun. 2014, 446, 1029–1034.
- Cai, J.; Bhatnagar, A.; Pierce, W.M. Protein modification by acrolein: Formation and stability of cysteine adducts. Chem. Res. Toxicol. 2009, 22, 708–716.
- Feng, Z.; Hu, W.; Hu, Y.; Tang, M. Acrolein is a major cigarette-related lung cancer agent: Preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc. Natl. Acad. Sci. USA 2006, 103, 15404–15409.
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12.
- Lugrin, J.; Rosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol. Chem. 2014, 395, 203–230.
- Xu, G.; Luo, K.; Liu, H.; Huang, T.; Fang, X.; Tu, W. The progress of inflammation and oxidative stress in patients with chronic kidney disease. Ren. Fail. 2015, 37, 45–49.
- Crawford, A.; Fassett, R.G.; Coombes, J.S.; Kunde, D.A.; Ahuja, K.D.K.; Robertson, I.K.; Ball, M.J.; Geraghty, D.P. Relationship between antioxidant enzyme genotype and activity and kidney function: A case-control study. Clin. Nephrol. 2012, 78, 135–144.
- Ling, X.C.; Kuo, K.-L. Oxidative stress in chronic kidney disease. Ren Replace Ther. 2018, 4, 53.
- Ceballos-Picot, I.; Witko-Sarsat, V.; Merad-Boudia, M.; Nguyen, A.T.; Thévenin, M.; Jaudon, M.C.; Zingraff, J.; Verger, C.; Jingers, P.; Descamps-Latscha, B. Glutathione antioxidant system as a marker of oxidative stress in chronic renal failure. Free Radic. Biol. Med. 1996, 21, 845–853.
- Small, D.M.; Gobe, G.C. Oxidative Stress and Antioxidant Therapy in Chronic Kidney and Cardiovascular Disease. In Oxidative Stress and Chronic Degenerative Diseases—A Role for Antioxidants; Morales-González, J.A., Ed.; IntechOpen: Rijeka, Croatia, 2013; ISBN 978-953-51-1123-8.
- Nishino, T.; Okamoto, K.; Kawaguchi, Y.; Hori, H.; Matsumura, T.; Eger, B.T.; Pai, E.F.; Nishino, T. Mechanism of the conversion of xanthine dehydrogenase to xanthine oxidase: Identification of the two cysteine disulfide bonds and crystal structure of a non-convertible rat liver xanthine dehydrogenase mutant. J. Biol. Chem. 2005, 280, 24888–24894.
- Liakopoulos, V.; Roumeliotis, S.; Gorny, X.; Dounousi, E.; Mertens, P.R. Oxidative Stress in Hemodialysis Patients: A Review of the Literature. Oxid. Med. Cell. Longev. 2017, 2017, 3081856.
- Cohen, G.; Hörl, W.H. Immune dysfunction in uremia—An update. Toxins 2012, 4, 962–990.
- Cosola, C.; Rocchetti, M.T.; Cupisti, A.; Gesualdo, L. Microbiota metabolites: Pivotal players of cardiovascular damage in chronic kidney disease. Pharmacol. Res. 2018, 130, 132–142.
- Saito, H.; Abe, T. (Eds.) Uremic Toxins and Organ Failure, 1st ed.; Springer: Singapore, 2020; ISBN 9789811577932.
- Wojtaszek, E.; Oldakowska-Jedynak, U.; Kwiatkowska, M.; Glogowski, T.; Malyszko, J. Uremic Toxins, Oxidative Stress, Atherosclerosis in Chronic Kidney Disease, and Kidney Transplantation. Oxid. Med. Cell. Longev. 2021, 2021, 6651367.
- Car, S.; Trkulja, V. Higher serum uric acid on admission is associated with higher short-term mortality and poorer long-term survival after myocardial infarction: Retrospective prognostic study. Croat. Med. J. 2009, 50, 559–566.
- Chen, J.-H.; Chuang, S.-Y.; Chen, H.-J.; Yeh, W.-T.; Pan, W.-H. Serum uric acid level as an independent risk factor for all-cause, cardiovascular, and ischemic stroke mortality: A Chinese cohort study. Arthritis Rheum. 2009, 61, 225–232.
- Letsas, K.P.; Korantzopoulos, P.; Filippatos, G.S.; Mihas, C.C.; Markou, V.; Gavrielatos, G.; Efremidis, M.; Sideris, A.; Kardaras, F. Uric acid elevation in atrial fibrillation. Hell. J. Cardiol. 2010, 51, 209–213.
- Alani, H.; Tamimi, A.; Tamimi, N. Cardiovascular co-morbidity in chronic kidney disease: Current knowledge and future research needs. World J. Nephrol. 2014, 3, 156–168.
- Tang, W.H.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584.
- Lichota, A.; Gwozdzinski, K.; Szewczyk, E.M. Microbial Modulation of Coagulation Disorders in Venous Thromboembolism. J. Inflamm. Res. 2020, 13, 387–400.
- Abou Dagher, G.; Harmouche, E.; Jabbour, E.; Bachir, R.; Zebian, D.; Bou Chebl, R. Sepsis in hemodialysis patients. BMC Emerg. Med. 2015, 15, 30.
- Pakhchanian, H.; Raiker, R.; Mukherjee, A.; Khan, A.; Singh, S.; Chatterjee, A. Outcomes of COVID-19 in CKD Patients: A Multicenter Electronic Medical Record Cohort Study. Clin. J. Am. Soc. Nephrol. 2021, 16, 785–786.


