 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Joy Yoon | + 2980 word(s) | 2980 | 2021-06-04 08:11:59 | | | |
| 2 | Peter Tang | Meta information modification | 2980 | 2021-06-25 04:17:39 | | |
Video Upload Options
The 1q21.1 CNVs, rare and large chromosomal microduplications and microdeletions, are detected in many patients with NDs. Phenotypes of duplication and deletion appear at the two ends of the spectrum. Microdeletions are predominant in individuals with schizophrenia (SCZ) and microcephaly, whereas microduplications are predominant in individuals with autism spectrum disorder (ASD) and macrocephaly.
1. Introduction
2. Chromosomal Mapping and Genetic Pathway of 1q21.1
2.1. Chromosomal Structure
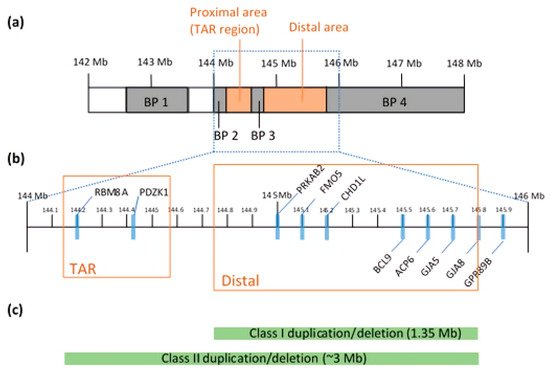
2.2. Genetic Architecture
|
Function 1 |
Molecular/Cellular Phenotypes |
References |
|
|---|---|---|---|
|
CHD1 |
Chromatin remodeling and DNA damage response |
Impaired decatenation checkpoint activation |
[25] |
|
PRKAB2 |
AMPK regulatory subunit; maintaining energy homeostasis |
Neurodegeneration; learning and memory impairment |
[42] |
|
GJA8 |
Gap junction protein; Connexin50 |
Cataracts; cardiac myopathy; increased risk of SCZ |
|
|
GJA5 |
Gap junction protein; Connexin40 |
Cataracts; cardiac abnormalities |
|
|
PDZK1 |
Ion transporter protein; regulates second messenger cascades |
Increased risk of ASD and psychosis |
[36] |
|
GPR89B |
Voltage dependent anion channel |
Unknown |
|
|
BCL9 |
Wnt signaling pathway |
Increased risk of SCZ |
[47] |
|
FMO5 |
Modulator of metabolic aging |
||
|
ACP6 |
Histidine acid phosphatase protein |
Unknown |
1 The Genecards Human Gene Database.
2.3. Pathogenesis of Proximal 1q21.1
3. Molecular and Cellular Mechanisms Associated with 1q21.1 CNVs
3.1. Effect Range of 1q21.1 CNVs
3.2. Synaptic Signaling Pathway
3.3. Mitochondrial Functions
3.4. The WNT Signaling Pathway and BCL9
References
- Girirajan, S.; Brkanac, Z.; Coe, B.P.; Baker, C.; Vives, L.; Vu, T.H.; Shafer, N.; Bernier, R.; Ferrero, G.B.; Silengo, M.; et al. Relative Burden of Large CNVs on a Range of Neurodevelopmental Phenotypes. PLoS Genet. 2011, 7, e1002334.
- Walsh, T.; McClellan, J.M.; McCarthy, S.E.; Addington, A.M.; Pierce, S.B.; Cooper, G.M.; Nord, A.S.; Kusenda, M.; Malhotra, D.; Bhandari, A.; et al. Rare Structural Variants Disrupt Multiple Genes in Neurodevelopmental Pathways in Schizophrenia. Science 2008, 320, 539–543.
- Kaminsky, E.B.; Kaul, V.; Paschall, J.; Church, D.M.; Bunke, B.; Kunig, D.; Moreno-De-Luca, D.; Moreno-De-Luca, A.; Mulle, J.G.; Warren, S.T.; et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet. Med. 2011, 13, 777–784.
- Girirajan, S.; Rosenfeld, J.A.; Coe, B.P.; Parikh, S.; Friedman, N.; Goldstein, A.; Filipink, R.A.; McConnell, J.S.; Angle, B.; Meschino, W.S.; et al. Phenotypic Heterogeneity of Genomic Disorders and Rare Copy-Number Variants. N. Engl. J. Med. 2012, 367, 1321–1331.
- Guyatt, A.L.; Stergiakouli, E.; Martin, J.; Walters, J.; O’Donovan, M.; Owen, M.; Thapar, A.; Kirov, G.; Rodriguez, S.; Rai, D.; et al. Association of copy number variation across the genome with neuropsychiatric traits in the general population. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2018, 177, 489–502.
- Coe, B.P.; Girirajan, S.; Eichler, E.E. The genetic variability and commonality of neurodevelopmental disease. Am. J. Med. Genet. Part C Semin. Med. Genet. 2012, 160C, 118–129.
- Forsingdal, A.; Jørgensen, T.N.; Olsen, L.; Werge, T.; Didriksen, M.; Nielsen, J. Can Animal Models of Copy Number Variants That Predispose to Schizophrenia Elucidate Underlying Biology? Biol. Psychiatry 2019, 85, 13–24.
- Stefansson, H.; Rujescu, D.; Cichon, S.; Pietiläinen, O.P.H.; Ingason, A.; Steinberg, S.; Fossdal, R.; Sigurdsson, E.; Sigmundsson, T.; Buizer-Voskamp, J.E.; et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008, 455, 232–236.
- Stone, J.L.; O’Donovan, M.C.; Gurling, H.; Kirov, G.K.; Blackwood, D.H.R.; Corvin, A.; Craddock, N.J.; Gill, M.; Hultman, C.M.; Lichtenstein, P.; et al. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008, 455, 237–241.
- Kirov, G.; Rees, E.; Walters, J.T.R.; Escott-Price, V.; Georgieva, L.; Richards, A.L.; Chambert, K.D.; Davies, G.; Legge, S.E.; Moran, J.L.; et al. The Penetrance of Copy Number Variations for Schizophrenia and Developmental Delay. Biol. Psychiatry 2014, 75, 378–385.
- Girirajan, S.; Rosenfeld, J.A.; Cooper, G.M.; Antonacci, F.; Siswara, P.; Itsara, A.; Vives, L.; Walsh, T.; McCarthy, S.E.; Baker, C.; et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010, 42, 203–209.
- Sriretnakumar, V.; Zai, C.C.; Wasim, S.; Barsanti-Innes, B.; Kennedy, J.L.; So, J. Copy number variant syndromes are frequent in schizophrenia: Progressing towards a CNV-schizophrenia model. Schizophr. Res. 2019, 209, 171–178.
- Rees, E.; Walters, J.T.R.; Georgieva, L.; Isles, A.R.; Chambert, K.D.; Richards, A.L.; Mahoney-Davies, G.; Legge, S.E.; Moran, J.L.; McCarroll, S.A.; et al. Analysis of copy number variations at 15 schizophrenia-associated loci. Br. J. Psychiatry 2014, 204, 108–114.
- Takumi, T.; Tamada, K. CNV biology in neurodevelopmental disorders. Curr. Opin. Neurobiol. 2018, 48, 183–192.
- Zhuo, C.; Hou, W.; Lin, C.; Hu, L.; Li, J. Potential Value of Genomic Copy Number Variations in Schizophrenia. Front. Mol. Neurosci. 2017, 10.
- Marshall, C.R.; Howrigan, D.P.; Merico, D.; Thiruvahindrapuram, B.; Wu, W.; Greer, D.S.; Antaki, D.; Shetty, A.; Holmans, P.A.; Pinto, D.; et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 2017, 49, 27–35.
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVs, including duplications of the 7q11. 23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885.
- Mefford, H.C.; Sharp, A.J.; Baker, C.; Itsara, A.; Jiang, Z.; Buysse, K.; Huang, S.; Maloney, V.K.; Crolla, J.A.; Baralle, D.; et al. Recurrent Rearrangements of Chromosome 1q21.1 and Variable Pediatric Phenotypes. N. Engl. J. Med. 2008, 359, 1685–1699.
- Sullivan, P.F. Schizophrenia and the dynamic genome. Genome Med. 2017, 9, 22.
- Malhotra, D.; Sebat, J. CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics. Cell 2012, 148, 1223–1241.
- Brunetti-Pierri, N.; Berg, J.S.; Scaglia, F.; Belmont, J.; Bacino, C.A.; Sahoo, T.; Lalani, S.R.; Graham, B.; Lee, B.; Shinawi, M.; et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008, 40, 1466–1471.
- Owen, M.J. Implications of Genetic Findings for Understanding Schizophrenia. Schizophr. Bull. 2012, 38, 904–907.
- Haldeman-Englert, C.; Jewett, T. 1q21.1 Microdeletion; University of Washington: Seattle, WA, USA, 1993–2021.
- Ripke, S.; O’Dushlaine, C.; Chambert, K.; Moran, J.L.; Kahler, A.K.; Akterin, S.; Bergen, S.E.; Collins, A.L.; Crowley, J.J.; Fromer, M.; et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013, 45, 1150–1159.
- Harvard, C.; Strong, E.; Mercier, E.; Colnaghi, R.; Alcantara, D.; Chow, E.; Martell, S.; Tyson, C.; Hrynchak, M.; McGillivray, B.; et al. Understanding the impact of 1q21.1 copy number variant. Orphanet. J. Rare Dis. 2011, 6, 54.
- Torres, F.; Barbosa, M.; Maciel, P. Recurrent copy number variations as risk factors for neurodevelopmental disorders: Critical overview and analysis of clinical implications. J. Med. Genet. 2016, 53, 73–90.
- Albers, C.A.; Paul, D.S.; Schulze, H.; Freson, K.; Stephens, J.C.; Smethurst, P.A.; Jolley, J.D.; Cvejic, A.; Kostadima, M.; Bertone, P.; et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat. Genet. 2012, 44, 435–439.
- Skorka, A.; Bielicka-Cymermann, J.; Gieruszczak-Bialek, D.; Korniszewski, L. Thrombocytopenia-absent radius (tar) syndrome: A case with agenesis of corpus callosum, hypoplasia of cerebellar vermis and horseshoe kidney. Genet. Couns. 2005, 16, 377–382.
- Weiss Sachdev, S.; Sunde, R.A. Selenium regulation of transcript abundance and translational efficiency of glutathione peroxidase-1 and -4 in rat liver. Biochem. J. 2001, 357, 851–858.
- Ceylan, A.C.; Sahin, I.; Erdem, H.B.; Kayhan, G.; Simsek-Kiper, P.O.; Utine, G.E.; Percin, F.; Boduroglu, K.; Alikasifoglu, M. An eight-case 1q21 region series: Novel aberrations and clinical variability with new features. J. Intellect. Disabil. Res. 2019, 63, 548–557.
- Mao, H.; Pilaz, L.-J.; McMahon, J.J.; Golzio, C.; Wu, D.; Shi, L.; Katsanis, N.; Silver, D.L. Rbm8a Haploinsufficiency Disrupts Embryonic Cortical Development Resulting in Microcephaly. J. Neurosci. 2015, 35, 7003–7018.
- Zou, D.; McSweeney, C.; Sebastian, A.; Reynolds, D.J.; Dong, F.; Zhou, Y.; Deng, D.; Wang, Y.; Liu, L.; Zhu, J.; et al. A critical role of RBM8a in proliferation and differentiation of embryonic neural progenitors. Neural. Dev. 2015, 10, 18.
- McSweeney, C.; Dong, F.; Chen, M.; Vitale, J.; Xu, L.; Crowley, N.; Luscher, B.; Zou, D.; Mao, Y. Full function of exon junction complex factor, Rbm8a, is critical for interneuron development. Transl. Psychiatry 2020, 10, 379.
- Alachkar, A.; Jiang, D.; Harrison, M.; Zhou, Y.; Chen, G.; Mao, Y. An EJC factor RBM8a Regulates Anxiety Behaviors. Curr. Mol. Med. 2013, 13, 887–899.
- Bernier, R.; Steinman, K.J.; Reilly, B.; Wallace, A.S.; Sherr, E.H.; Pojman, N.; Mefford, H.C.; Gerdts, J.; Earl, R.; Hanson, E.; et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet. Med. 2016, 18, 341–349.
- Busè, M.; Cuttaia, H.C.; Palazzo, D.; Mazara, M.V.; Lauricella, S.A.; Malacarne, M.; Pierluigi, M.; Cavani, S.; Piccione, M. Expanding the phenotype of reciprocal 1q21.1 deletions and duplications: A case series. Ital. J. Pediatrics 2017, 43, 61.
- Golzio, C.; Katsanis, N. Genetic architecture of reciprocal CNVs. Curr. Opin. Genet. Dev. 2013, 23, 240–248.
- Hall, J.; Trent, S.; Thomas, K.L.; O’Donovan, M.C.; Owen, M.J. Genetic Risk for Schizophrenia: Convergence on Synaptic Pathways Involved in Plasticity. Biol. Psychiatry 2015, 77, 52–58.
- Deshpande, A.; Weiss, L.A. Recurrent reciprocal copy number variants: Roles and rules in neurodevelopmental disorders. Dev. Neurobiol. 2018, 78, 519–530.
- Crespi, B.J.; Crofts, H.J. Association testing of copy number variants in schizophrenia and autism spectrum disorders. J. Neurodev. Disord. 2012, 4, 15.
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220.
- Nagy, S.; Maurer, G.W.; Hentze, J.L.; Rose, M.; Werge, T.M.; Rewitz, K. AMPK signaling linked to the schizophrenia-associated 1q21.1 deletion is required for neuronal and sleep maintenance. PLoS Genet. 2018, 14, e1007623.
- Rong, P.; Wang, X.; Niesman, I.; Wu, Y.; Benedetti, L.E.; Dunia, I.; Levy, E.; Gong, X. Disruption of ja8 (α8 connexin) in mice leads to microphthalmia associated with retardation of lens growth and lens fiber maturation. Development 2002, 129, 167–174.
- Ni, X.; Valente, J.; Azevedo, M.H.; Pato, M.T.; Pato, C.N.; Kennedy, J.L. Connexin 50 gene on human chromosome 1q21 is associated with schizophrenia in matched case–control and family-based studies. J. Med Genet. 2007, 44, 532–536.
- Verhagen, J.M.A.; de Leeuw, N.; Papatsonis, D.N.M.; Grijseels, E.W.M.; de Krijger, R.R.; Wessels, M.W. Phenotypic Variability Associated with a Large Recurrent 1q21.1 Microduplication in a Three-Generation Family. Mol. Syndromol. 2015, 6, 71–76.
- Simon, A.M.; Goodenough, D.A.; Paul, D.L. Mice lacking connexin40 have cardiac conduction abnormalities characteristic of atrioventricular block and bundle branch block. Curr. Biol. 1998, 8, 295–298.
- Li, J.; Zhou, G.; Ji, W.; Feng, G.; Zhao, Q.; Liu, J.; Li, T.; Li, Y.; Chen, P.; Zeng, Z.; et al. Common Variants in the BCL9 Gene Conferring Risk of Schizophrenia. Arch. Gen. Psychiatry 2011, 68, 232–240.
- Varshavi, D.; Scott, F.H.; Varshavi, D.; Veeravalli, S.; Phillips, I.R.; Veselkov, K.; Strittmatter, N.; Takats, Z.; Shephard, E.A.; Everett, J.R. Metabolic Biomarkers of Ageing in C57BL/6J Wild-Type and Flavin-Containing Monooxygenase 5 (FMO5)-Knockout Mice. Front. Mol. Biosci. 2018, 5, 28.
- Gagliardi, S.; Abel, K.; Bianchi, M.; Milani, P.; Bernuzzi, S.; Corato, M.; Ceroni, M.; Cashman, J.R.; Cereda, C. Regulation of FMO and PON Detoxication Systems in ALS Human Tissues. Neurotox. Res. 2013, 23, 370–377.
- Pang, H.; Yu, X.; Kim, Y.M.; Wang, X.; Jinkins, J.K.; Yin, J.; Li, S.; Gu, H. Disorders Associated With Diverse, Recurrent Deletions and Duplications at 1q21.1. Front. Genet. 2020, 11.
- Rosenfeld, J.A.; Traylor, R.N.; Schaefer, G.B.; McPherson, E.W.; Ballif, B.C.; Klopocki, E.; Mundlos, S.; Shaffer, L.G.; Aylsworth, A.S. Proximal microdeletions and microduplications of 1q21.1 contribute to variable abnormal phenotypes. Eur. J. Hum. Genet. 2012, 20, 754–761.
- Rees, E.; Moskvina, V.; Owen, M.J.; O’Donovan, M.C.; Kirov, G. De Novo Rates and Selection of Schizophrenia-Associated Copy Number Variants. Biol. Psychiatry 2011, 70, 1109–1114.
- Kirov, G.; Pocklington, A.J.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 2012, 17, 142–153.
- Reinwald, J.R.; Sartorius, A.; Weber-Fahr, W.; Sack, M.; Becker, R.; Didriksen, M.; Stensbøl, T.B.; Schwarz, A.J.; Meyer-Lindenberg, A.; Gass, N. Separable neural mechanisms for the pleiotropic association of copy number variants with neuropsychiatric traits. Transl. Psychiatry 2020, 10, 93.
- Nielsen, J.; Fejgin, K.; Sotty, F.; Nielsen, V.; Mørk, A.; Christoffersen, C.T.; Yavich, L.; Lauridsen, J.B.; Clausen, D.; Larsen, P.H.; et al. A mouse model of the schizophrenia-associated 1q21.1 microdeletion syndrome exhibits altered mesolimbic dopamine transmission. Transl. Psychiatry 2017, 7, 1261.
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233.
- Marotta, R.; Risoleo, M.C.; Messina, G.; Parisi, L.; Carotenuto, M.; Vetri, L.; Roccella, M. The Neurochemistry of Autism. Brain Sci. 2020, 10, 163.
- Howes, O.D.; Kambeitz, J.; Kim, E.; Stahl, D.; Slifstein, M.; Abi-Dargham, A.; Kapur, S. The Nature of Dopamine Dysfunction in Schizophrenia and What This Means for Treatment: Meta-analysis of Imaging Studies. Arch. Gen. Psychiatry 2012, 69, 776–786.
- Howes, O.D.; Shotbolt, P.; Bloomfield, M.; Daalman, K.; Demjaha, A.; Diederen, K.M.J.; Ibrahim, K.; Kim, E.; McGuire, P.; Kahn, R.S.; et al. Dopaminergic Function in the Psychosis Spectrum: An [18F]-DOPA Imaging Study in Healthy Individuals with Auditory Hallucinations. Schizophr. Bull. 2013, 39, 807–814.
- Liu, Z.; Osipovitch, M.; Benraiss, A.; Huynh, N.P.T.; Foti, R.; Bates, J.; Chandler-Militello, D.; Findling, R.L.; Tesar, P.J.; Nedergaard, M.; et al. Dysregulated Glial Differentiation in Schizophrenia May Be Relieved by Suppression of SMAD4- and REST-Dependent Signaling. Cell Rep. 2019, 27, 3832–3843.e3836.
- Dror, V.; Shamir, E.; Ghanshani, S.; Kimhi, R.; Swartz, M.; Barak, Y.; Weizman, R.; Avivi, L.; Litmanovitch, T.; Fantino, E.; et al. hKCa3/KCNN3 potassium channel gene: Association of longer CAG repeats with schizophrenia in Israeli Ashkenazi Jews, expression in human tissues and localization to chromosome 1q21. Mol. Psychiatry 1999, 4, 254–260.
- Austin, C.P.; Holder, D.J.; Ma, L.; Mixson, L.A.; Caskey, C.T. Mapping of hKCa3 to chromosome 1q21 and investigation of linkage of CAG repeat polymorphism to schizophrenia. Mol. Psychiatry 1999, 4, 261–266.
- Meissner, B.; Purmann, S.; Schürmann, M.; Zühlke, C.; Lencer, R.; Arolt, V.; Müller-Myhsok, B.; Morris-Rosendahl, D.J.; Schwinger, E. hSKCa3: A candidate gene for schizophrenia? Psychiatr. Genet. 1999, 9, 91–96.
- Miller, M.J.; Rauer, H.; Tomita, H.; Rauer, H.; Gargus, J.J.; Gutman, G.A.; Cahalan, M.D.; Chandy, K.G. Nuclear Localization and Dominant-negative Suppression by a Mutant SKCa3 N-terminal Channel Fragment Identified in a Patient with Schizophrenia. J. Biol. Chem. 2001, 276, 27753–27756.
- Frye, R.E.; Vassall, S.; Kaur, G.; Lewis, C.; Karim, M.; Rossignol, D. Emerging biomarkers in autism spectrum disorder: A systematic review. Ann. Transl. Med. 2019, 7, 792.
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314.
- Ng, C.-H.; Guan, M.S.H.; Koh, C.; Ouyang, X.; Yu, F.; Tan, E.-K.; O’Neill, S.P.; Zhang, X.; Chung, J.; Lim, K.-L. AMP Kinase Activation Mitigates Dopaminergic Dysfunction and Mitochondrial Abnormalities in Drosophila Models of Parkinson’s Disease. J. Neurosci. 2012, 32, 14311–14317.
- Gordon, A.; Forsingdal, A.; Klewe, I.V.; Nielsen, J.; Didriksen, M.; Werge, T.; Geschwind, D.H. Transcriptomic networks implicate neuronal energetic abnormalities in three mouse models harboring autism and schizophrenia-associated mutations. Mol. Psychiatry 2019.
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.-J.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 2004, 9, 684–697.
- Roberts, R.C. Mitochondrial dysfunction in schizophrenia: With a focus on postmortem studies. Mitochondrion 2021, 56, 91–101.
- Sunyer, J.; Dadvand, P. Pre-natal brain development as a target for urban air pollution. Basic Clin. Pharmacol. Toxicol. 2019, 125, 81–88.
- Windham, G.C.; Pearl, M.; Poon, V.; Berger, K.; Soriano, J.W.; Eyles, D.; Lyall, K.; Kharrazi, M.; Croen, L.A. Maternal Vitamin D Levels During Pregnancy in Association With Autism Spectrum Disorders (ASD) or Intellectual Disability (ID) in Offspring; Exploring Non-linear Patterns and Demographic Sub-groups. Autism. Res. 2020, 13, 2216–2229.
- Jones, H.F.; Ho, A.C.C.; Sharma, S.; Mohammad, S.S.; Kothur, K.; Patel, S.; Brilot, F.; Guastella, A.J.; Dale, R.C.; Group, I.-N.S. Maternal thyroid autoimmunity associated with acute-onset neuropsychiatric disorders and global regression in offspring. Dev. Med. Child Neurol. 2019, 61, 984–988.
- Van den Bergh, B.R.H.; van den Heuvel, M.I.; Lahti, M.; Braeken, M.; de Rooij, S.R.; Entringer, S.; Hoyer, D.; Roseboom, T.; Räikkönen, K.; King, S.; et al. Prenatal developmental origins of behavior and mental health: The influence of maternal stress in pregnancy. Neurosci. Biobehav. Rev. 2020, 117, 26–64.
- Webb, S.J.; Garrison, M.M.; Bernier, R.; McClintic, A.M.; King, B.H.; Mourad, P.D. Severity of ASD symptoms and their correlation with the presence of copy number variations and exposure to first trimester ultrasound. Autism Res. 2017, 10, 472–484.
- Shelton, J.F.; Geraghty, E.M.; Tancredi, D.J.; Delwiche, L.D.; Schmidt, R.J.; Ritz, B.; Hansen, R.L.; Hertz-Picciotto, I. Neurodevelopmental Disorders and Prenatal Residential Proximity to Agricultural Pesticides: The CHARGE Study. Environ. Health Perspect. 2014, 122, 1103–1109.
- Pedersen, M.G.; Stevens, H.; Pedersen, C.B.; Norgaard-Pedersen, B.; Mortensen, P.B. Toxoplasma infection and later development of schizophrenia in mothers. Am. J. Psychiatry 2011, 168, 814–821.
- Dickerson, F.; Kirkpatrick, B.; Boronow, J.; Stallings, C.; Origoni, A.; Yolken, R. Deficit schizophrenia: Association with serum antibodies to cytomegalovirus. Schizophr. Bull. 2006, 32, 396–400.
- O’Callaghan, E.; Sham, P.; Takei, N.; Glover, G.; Murray, R.M. Schizophrenia after prenatal exposure to 1957 A2 influenza epidemic. Lancet 1991, 337, 1248–1250.
- Suvisaari, J.; Haukka, J.; Tanskanen, A.; Hovi, T.; Lonnqvist, J. Association between prenatal exposure to poliovirus infection and adult schizophrenia. Am. J. Psychiatry 1999, 156, 1100–1102.
- Susser, E.S.; Lin, S.P. Schizophrenia after prenatal exposure to the Dutch Hunger Winter of 1944–1945. Arch. Gen. Psychiatry 1992, 49, 983–988.
- St Clair, D.; Xu, M.; Wang, P.; Yu, Y.; Fang, Y.; Zhang, F.; Zheng, X.; Gu, N.; Feng, G.; Sham, P.; et al. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959–1961. JAMA 2005, 294, 557–562.
- Shen, Q.; Li, Z.Q.; Sun, Y.; Wang, T.; Wan, C.L.; Li, X.W.; Zhao, X.Z.; Feng, G.Y.; Li, S.; St Clair, D.; et al. The role of pro-inflammatory factors in mediating the effects on the fetus of prenatal undernutrition: Implications for schizophrenia. Schizophr. Res. 2008, 99, 48–55.
- O’Donnell, K.; O’Connor, T.G.; Glover, V. Prenatal stress and neurodevelopment of the child: Focus on the HPA axis and role of the placenta. Dev. Neurosci. 2009, 31, 285–292.
- Jablensky, A.V.; Morgan, V.; Zubrick, S.R.; Bower, C.; Yellachich, L.A. Pregnancy, delivery, and neonatal complications in a population cohort of women with schizophrenia and major affective disorders. Am. J. Psychiatry 2005, 162, 79–91.
- Gradinaru, V.; Zhang, F.; Ramakrishnan, C.; Mattis, J.; Prakash, R.; Diester, I.; Goshen, I.; Thompson, K.R.; Deisseroth, K. Molecular and cellular approaches for diversifying and extending optogenetics. Cell 2010, 141, 154–165.
- Junge, H.J.; Yang, S.; Burton, J.B.; Paes, K.; Shu, X.; French, D.M.; Costa, M.; Rice, D.S.; Ye, W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell 2009, 139, 299–311.
- Ye, X.; Wang, Y.; Cahill, H.; Yu, M.; Badea, T.C.; Smallwood, P.M.; Peachey, N.S.; Nathans, J. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell 2009, 139, 285–298.
- Phng, L.K.; Potente, M.; Leslie, J.D.; Babbage, J.; Nyqvist, D.; Lobov, I.; Ondr, J.K.; Rao, S.; Lang, R.A.; Thurston, G.; et al. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev. Cell 2009, 16, 70–82.
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 641–646.
- Kalani, M.Y.; Cheshier, S.H.; Cord, B.J.; Bababeygy, S.R.; Vogel, H.; Weissman, I.L.; Palmer, T.D.; Nusse, R. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16970–16975.
- Viti, J.; Gulacsi, A.; Lillien, L. Wnt regulation of progenitor maturation in the cortex depends on Shh or fibroblast growth factor 2. J. Neurosci. 2003, 23, 5919–5927.
- Lyu, J.; Joo, C.K. Wnt signaling enhances FGF2-triggered lens fiber cell differentiation. Development 2004, 131, 1813–1824.
- Kuwabara, T.; Hsieh, J.; Muotri, A.; Yeo, G.; Warashina, M.; Lie, D.C.; Moore, L.; Nakashima, K.; Asashima, M.; Gage, F.H. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 2009, 12, 1097–1105.
- Munji, R.N.; Choe, Y.; Li, G.; Siegenthaler, J.A.; Pleasure, S.J. Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J. Neurosci. 2011, 31, 1676–1687.
- Wang, Q.; Charych, E.I.; Pulito, V.L.; Lee, J.B.; Graziane, N.M.; Crozier, R.A.; Revilla-Sanchez, R.; Kelly, M.P.; Dunlop, A.J.; Murdoch, H.; et al. The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function. Mol. Psychiatry 2011, 16, 1006–1023.
- De Rienzo, G.; Bishop, J.A.; Mao, Y.; Pan, L.; Ma, T.P.; Moens, C.B.; Tsai, L.H.; Sive, H. Disc1 regulates both beta-catenin-mediated and noncanonical Wnt signaling during vertebrate embryogenesis. FASEB J. 2011, 25, 4184–4197.
- Lie, D.C.; Colamarino, S.A.; Song, H.J.; Desire, L.; Mira, H.; Consiglio, A.; Lein, E.S.; Jessberger, S.; Lansford, H.; Dearie, A.R.; et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature 2005, 437, 1370–1375.
- L’Episcopo, F.; Serapide, M.F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Pluchino, S.; Marchetti, B. A Wnt1 regulated Frizzled-1/beta-Catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: Therapeutical relevance for neuron survival and neuroprotection. Mol. Neurodegener. 2011, 6, 49.
- Faulkner, R.L.; Jang, M.H.; Liu, X.B.; Duan, X.; Sailor, K.A.; Kim, J.Y.; Ge, S.; Jones, E.G.; Ming, G.L.; Song, H.; et al. Development of hippocampal mossy fiber synaptic outputs by new neurons in the adult brain. Proc. Natl. Acad. Sci. USA 2008, 105, 14157–14162.
- Alvarez, A.R.; Godoy, J.A.; Mullendorff, K.; Olivares, G.H.; Bronfman, M.; Inestrosa, N.C. Wnt-3a overcomes beta-amyloid toxicity in rat hippocampal neurons. Exp. Cell Res. 2004, 297, 186–196.
- Chong, Z.Z.; Maiese, K. Targeting WNT, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system. Histol. Histopathol. 2004, 19, 495–504.
- Sahores, M.; Gibb, A.; Salinas, P.C. Frizzled-5, a receptor for the synaptic organizer Wnt7a, regulates activity-mediated synaptogenesis. Development 2010, 137, 2215–2225.
- Klassen, M.P.; Shen, K. Wnt signaling positions neuromuscular connectivity by inhibiting synapse formation in C. elegans. Cell 2007, 130, 704–716.
- Hall, A.C.; Lucas, F.R.; Salinas, P.C. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell 2000, 100, 525–535.
- Krylova, O.; Messenger, M.J.; Salinas, P.C. Dishevelled-1 regulates microtubule stability: A new function mediated by glycogen synthase kinase-3beta. J. Cell Biol. 2000, 151, 83–94.
- Lyuksyutova, A.I.; Lu, C.C.; Milanesio, N.; King, L.A.; Guo, N.; Wang, Y.; Nathans, J.; Tessier-Lavigne, M.; Zou, Y. Anterior-posterior guidance of commissural axons by Wnt-frizzled signaling. Science 2003, 302, 1984–1988.
- Zechner, D.; Fujita, Y.; Hülsken, J.; Müller, T.; Walther, I.; Taketo, M.M.; Crenshaw, I.I.I.E.B.; Birchmeier, W.; Birchmeier, C. β-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev. Biol. 2003, 258, 406–418.
- De Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929.
- Kuechler, A.; Willemsen, M.H.; Albrecht, B.; Bacino, C.A.; Bartholomew, D.W.; van Bokhoven, H.; van den Boogaard, M.J.; Bramswig, N.; Buttner, C.; Cremer, K.; et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: Expanding the mutational and clinical spectrum. Hum. Genet. 2015, 134, 97–109.
- Winczewska-Wiktor, A.; Badura-Stronka, M.; Monies-Nowicka, A.; Nowicki, M.M.; Steinborn, B.; Latos-Bielenska, A.; Monies, D. A de novo CTNNB1 nonsense mutation associated with syndromic atypical hyperekplexia, microcephaly and intellectual disability: A case report. BMC Neurol. 2016, 16, 35.
- Kharbanda, M.; Pilz, D.T.; Tomkins, S.; Chandler, K.; Saggar, A.; Fryer, A.; McKay, V.; Louro, P.; Smith, J.C.; Burn, J.; et al. Clinical features associated with CTNNB1 de novo loss of function mutations in ten individuals. Eur. J. Med Genet. 2017, 60, 130–135.
- Dubruc, E.; Putoux, A.; Labalme, A.; Rougeot, C.; Sanlaville, D.; Edery, P. A new intellectual disability syndrome caused by CTNNB1 haploinsufficiency. Am. J. Med. Genet. Part A 2014, 164A, 1571–1575.
- Tucci, V.; Kleefstra, T.; Hardy, A.; Heise, I.; Maggi, S.; Willemsen, M.H.; Hilton, H.; Esapa, C.; Simon, M.; Buenavista, M.T.; et al. Dominant beta-catenin mutations cause intellectual disability with recognizable syndromic features. J. Clin. Investig. 2014, 124, 1468–1482.
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250.
- Stessman, H.A.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526.
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science 2012.
- Iakoucheva, L.M.; Muotri, A.R.; Sebat, J. Getting to the Cores of Autism. Cell 2019, 178, 1287–1298.
- Krupp, D.R.; Barnard, R.A.; Duffourd, Y.; Evans, S.A.; Mulqueen, R.M.; Bernier, R.; Rivière, J.-B.; Fombonne, E.; O’Roak, B.J. Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am. J. Hum. Genet. 2017, 101, 369–390.
- Dong, F.; Jiang, J.; McSweeney, C.; Zou, D.; Liu, L.; Mao, Y. Deletion of CTNNB1 in inhibitory circuitry contributes to autism-associated behavioral defects. Hum. Mol. Genet. 2016, 25, 2738–2751.
- Mohn, J.L.; Alexander, J.; Pirone, A.; Palka, C.D.; Lee, S.Y.; Mebane, L.; Haydon, P.G.; Jacob, M.H. Adenomatous polyposis coli protein deletion leads to cognitive and autism-like disabilities. Mol. Psychiatry 2014, 19, 1133–1142.
- Lijam, N.; Paylor, R.; McDonald, M.P.; Crawley, J.N.; Deng, C.-X.; Herrup, K.; Stevens, K.E.; Maccaferri, G.; McBain, C.J.; Sussman, D.J.; et al. Social Interaction and Sensorimotor Gating Abnormalities in Mice Lacking Dvl1. Cell 1997, 90, 895–905.
- Shin, S.; Santi, A.; Huang, S. Conditional Pten knockout in parvalbumin- or somatostatin-positive neurons sufficiently leads to autism-related behavioral phenotypes. Mol. Brain 2021, 14, 24.
- Busch, R.M.; Srivastava, S.; Hogue, O.; Frazier, T.W.; Klaas, P.; Hardan, A.; Martinez-Agosto, J.A.; Sahin, M.; Eng, C. Neurobehavioral phenotype of autism spectrum disorder associated with germline heterozygous mutations in PTEN. Transl. Psychiatry 2019, 9, 253.
- Chen, C.-J.; Sgritta, M.; Mays, J.; Zhou, H.; Lucero, R.; Park, J.; Wang, I.C.; Park, J.H.; Kaipparettu, B.A.; Stoica, L.; et al. Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat. Med. 2019, 25, 1684–1690.
- Lugo, J.N.; Smith, G.D.; Arbuckle, E.P.; White, J.; Holley, A.J.; Floruta, C.M.; Ahmed, N.; Gomez, M.C.; Okonkwo, O. Deletion of PTEN produces autism-like behavioral deficits and alterations in synaptic proteins. Front. Mol. Neurosci. 2014, 7.
- Kramps, T.; Peter, O.; Brunner, E.; Nellen, D.; Froesch, B.; Chatterjee, S.; Murone, M.; Züllig, S.; Basler, K. Wnt/Wingless Signaling Requires BCL9/Legless-Mediated Recruitment of Pygopus to the Nuclear β-Catenin-TCF Complex. Cell 2002, 109, 47–60.
- Brembeck, F.H.; Schwarz-Romond, T.; Bakkers, J.; Wilhelm, S.; Hammerschmidt, M.; Birchmeier, W. Essential role of BCL9-2 in the switch between β-catenin’s adhesive and transcriptional functions. Genes Dev. 2004, 18, 2225–2230.
- Mieszczanek, J.; de la Roche, M.; Bienz, M. A role of Pygopus as an anti-repressor in facilitating Wnt-dependent transcription. Proc. Natl. Acad. Sci. USA 2008, 105, 19324–19329.
- Brack, A.S.; Murphy-Seiler, F.; Hanifi, J.; Deka, J.; Eyckerman, S.; Keller, C.; Aguet, M.; Rando, T.A. BCL9 is an essential component of canonical Wnt signaling that mediates the differentiation of myogenic progenitors during muscle regeneration. Dev. Biol. 2009, 335, 93–105.
- Xu, C.; Aragam, N.; Li, X.; Villla, E.C.; Wang, L.; Briones, D.; Petty, L.; Posada, Y.; Arana, T.B.; Cruz, G.; et al. BCL9 and C9orf5 Are Associated with Negative Symptoms in Schizophrenia: Meta-Analysis of Two Genome-Wide Association Studies. PLoS ONE 2013, 8, e51674.
- Kimura, H.; Tanaka, S.; Kushima, I.; Koide, T.; Banno, M.; Kikuchi, T.; Nakamura, Y.; Shiino, T.; Yoshimi, A.; Oya-Ito, T.; et al. Association study of BCL9 gene polymorphism rs583583 with schizophrenia and negative symptoms in Japanese population. Sci. Rep. 2015, 5, 15705.
- Luo, X.; Huang, L.; Han, L.; Luo, Z.; Hu, F.; Tieu, R.; Gan, L. Systematic Prioritization and Integrative Analysis of Copy Number Variations in Schizophrenia Reveal Key Schizophrenia Susceptibility Genes. Schizophr. Bull. 2014, 40, 1285–1299.
- Takada, K.; Zhu, D.; Bird, G.H.; Sukhdeo, K.; Zhao, J.-J.; Mani, M.; Lemieux, M.; Carrasco, D.E.; Ryan, J.; Horst, D.; et al. Targeted Disruption of the BCL9/β-Catenin Complex Inhibits Oncogenic Wnt Signaling. Sci. Transl. Med. 2012, 4, 148ra117.
- Moor, A.E.; Anderle, P.; Cantù, C.; Rodriguez, P.; Wiedemann, N.; Baruthio, F.; Deka, J.; André, S.; Valenta, T.; Moor, M.B.; et al. BCL9/9L-β-catenin Signaling is Associated With Poor Outcome in Colorectal Cancer. EBioMedicine 2015, 2, 1932–1943.
- Beaulieu, J.-F. Tuning WNT-β-catenin signaling via BCL9 proteins for targeting colorectal cancer cells. EBioMedicine 2015, 2, 1846–1847.
- Miyaoka, T.; Seno, H.; Ishino, H. Increased expression of Wnt-1 in schizophrenic brains. Schizophr. Res. 1999, 38, 1–6.
- Proitsi, P.; Li, T.; Hamilton, G.; Di Forti, M.; Collier, D.; Killick, R.; Chen, R.; Sham, P.; Murray, R.; Powell, J.; et al. Positional pathway screen of wnt signaling genes in schizophrenia: Association with DKK4. Biol. Psychiatry 2008, 63, 13–16.
- Yang, J.; Si, T.; Ling, Y.; Ruan, Y.; Han, Y.; Wang, X.; Zhang, H.; Kong, Q.; Li, X.; Liu, C.; et al. Association study of the human FZD3 locus with schizophrenia. Biol. Psychiatry 2003, 54, 1298–1301.
- Ide, M.; Muratake, T.; Yamada, K.; Iwayama-Shigeno, Y.; Iwamoto, K.; Takao, H.; Toyota, T.; Kaneko, N.; Minabe, Y.; Nakamura, K.; et al. Genetic and expression analyses of FZD3 in schizophrenia. Biol. Psychiatry 2004, 56, 462–465.
- Zhang, Y.; Yu, X.; Yuan, Y.; Ling, Y.; Ruan, Y.; Si, T.; Lu, T.; Wu, S.; Gong, X.; Zhu, Z.; et al. Positive association of the human frizzled 3 (FZD3) gene haplotype with schizophrenia in Chinese Han population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2004, 129, 16–19.
- Lachman, H.M.; Pedrosa, E.; Petruolo, O.A.; Cockerham, M.; Papolos, A.; Novak, T.; Papolos, D.F.; Stopkova, P. Increase in GSK3beta gene copy number variation in bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144, 259–265.
- Martin, P.M.; Yang, X.; Robin, N.; Lam, E.; Rabinowitz, J.S.; Erdman, C.A.; Quinn, J.; Weiss, L.A.; Hamilton, S.P.; Kwok, P.Y.; et al. A rare WNT1 missense variant overrepresented in ASD leads to increased Wnt signal pathway activation. Transl. Psychiatry 2013, 3, e301.
- Millar, J.K.; Wilson-Annan, J.C.; Anderson, S.; Christie, S.; Taylor, M.S.; Semple, C.A.; Devon, R.S.; Clair, D.M.; Muir, W.J.; Blackwood, D.H.; et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 2000, 9, 1415–1423.
- Kim, H.J.; Park, H.J.; Jung, K.H.; Ban, J.Y.; Ra, J.; Kim, J.W.; Park, J.K.; Choe, B.K.; Yim, S.V.; Kwon, Y.K.; et al. Association study of polymorphisms between DISC1 and schizophrenia in a Korean population. Neurosci. Lett. 2007, 26, 81–96.
- Tomppo, L.; Hennah, W.; Lahermo, P.; Loukola, A.; Tuulio-Henriksson, A.; Suvisaari, J.; Partonen, T.; Ekelund, J.; Lonnqvist, J.; Peltonen, L. Association between genes of Disrupted in schizophrenia 1 (DISC1) interactors and schizophrenia supports the role of the DISC1 pathway in the etiology of major mental illnesses. Biol. Psychiatry 2009, 65, 1055–1062.
- Schosser, A.; Gaysina, D.; Cohen-Woods, S.; Chow, P.C.; Martucci, L.; Craddock, N.; Farmer, A.; Korszun, A.; Gunasinghe, C.; Gray, J.; et al. Association of DISC1 and TSNAX genes and affective disorders in the depression case-control (DeCC) and bipolar affective case-control (BACCS) studies. Mol. Psychiatry 2009.
- Hennah, W.; Thomson, P.; McQuillin, A.; Bass, N.; Loukola, A.; Anjorin, A.; Blackwood, D.; Curtis, D.; Deary, I.J.; Harris, S.E.; et al. DISC1 association, heterogeneity and interplay in schizophrenia and bipolar disorder. Mol. Psychiatry 2009, 14, 865–873.
- Kilpinen, H.; Ylisaukko-Oja, T.; Hennah, W.; Palo, O.M.; Varilo, T.; Vanhala, R.; Nieminen-von Wendt, T.; von Wendt, L.; Paunio, T.; Peltonen, L. Association of DISC1 with autism and Asperger syndrome. Mol. Psychiatry 2007, 13, 187–196.
- Mao, Y.; Ge, X.; Frank, C.L.; Madison, J.M.; Koehler, A.N.; Doud, M.K.; Tassa, C.; Berry, E.M.; Soda, T.; Singh, K.K.; et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 2009, 136, 1017–1031.


