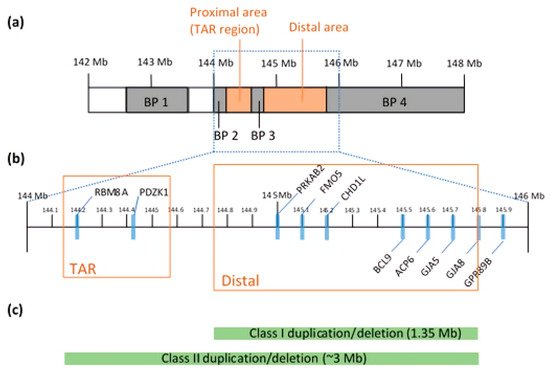
The 1q21.1 CNV is found within a 144 to 148 Mb region
[18] (a). In contrast to small CNVs, which are less detrimental, larger CNVs (>500 kb in size) can alter the expression levels of multiple genes
[19]. It is a complex locus to study in that it not only spans 20–40 putative genes, but the region is also susceptible to genomic rearrangements due to the numbers of low copy repeats (LCRs). The more LCRs in the region, the more prone it is to frequent non-allelic homologous recombination (NAHR) during meiosis
[20]. Clustered with LCRs, breakpoints (BPs) divide the locus into four possible segmental blocks and complicate the mapping and prediction of phenotypic expressivity
[18]. Many of the LCRs and BPs are located adjacent to the crossing over points, making it difficult to estimate the phenotypes or genomic sequences in any given persons
[21]. Through this mechanism, the CNVs, emerging in chromosomal duplications or deletions, can alter some of the dosage-sensitive genes and create a broad range of phenotypic variability
[22]. Array comparative genomic hybridization and fluorescent in-situ hybridization analyses mapped out the overall structure of the 1q21.1 in great detail. The 1q21.1 region is associated with mental retardation, autism
[23], schizophrenia
[24] and microcephaly
[21]. Duplication of 1q21.1 is strongly associated with autism
[21].
Duplications and deletions are classified into two classes: Class I and Class II. Class I duplication/deletion involves only the distal 1q21.1 region between BP3 and BP4 (1.35 Mb in size), whereas Class II duplication/deletion extends from the distal 1q21.1 to the proximal 1q21.1 commonly detected between BP2 and BP4 (~3 Mb)
[21] (c). Combined data show enrichment in Class I deletions and duplications with a parental origin, but the components of genes and BPs can be varied after generations
[25]. Both analyses discovered two distinct regions: proximal and distal 1q21.1, where a genomic gain or loss occurs (b)
[21][26][21,26]. Microdeletions at proximal 1q21.1 are mainly associated with thrombocytopenia-absent radius (TAR) syndrome and this region is often referred to as the TAR region. In particular, a core exon junction complex gene,
RBM8A, is located in the TAR region and compound mutations in the
RBM8A gene cause the TAR syndrome
[27] that is comorbid with ID
[28]. Other brain dysfunctions, including psychosis, agenesis of the corpus callosum and hypoplasia of the cerebellar vermis, are present in TAR patients
[28][29][30][28,29,30]. Consistent with human patient studies, knockdown and knockout of
Rbm8a in a mouse model revealed the critical role of
RBM8A in neural progenitor cell (NPC) proliferation, neuronal migration and interneuron development, and loss of function in
RBM8A in NPCs causes microcephaly
[31][32][33][31,32,33]. Moreover,
RBM8A plays a key role in adult neurogenesis and in regulating anxiety-related behavior
[34], further supporting the important role of
RBM8A in psychiatric diseases.