Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mina Lobbous | + 2755 word(s) | 2755 | 2021-06-11 05:10:45 | | | |
| 2 | Vivi Li | Meta information modification | 2755 | 2021-06-25 08:20:03 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lobbous, M. Neurofibromatosis Type 1-Associated Gliomas. Encyclopedia. Available online: https://encyclopedia.pub/entry/11279 (accessed on 07 February 2026).
Lobbous M. Neurofibromatosis Type 1-Associated Gliomas. Encyclopedia. Available at: https://encyclopedia.pub/entry/11279. Accessed February 07, 2026.
Lobbous, Mina. "Neurofibromatosis Type 1-Associated Gliomas" Encyclopedia, https://encyclopedia.pub/entry/11279 (accessed February 07, 2026).
Lobbous, M. (2021, June 24). Neurofibromatosis Type 1-Associated Gliomas. In Encyclopedia. https://encyclopedia.pub/entry/11279
Lobbous, Mina. "Neurofibromatosis Type 1-Associated Gliomas." Encyclopedia. Web. 24 June, 2021.
Copy Citation
Neurofibromatosis type 1 (NF1) is an autosomal dominant tumor predisposition syndrome that affects children and adults. Individuals with NF1 are at high risk for central nervous system neoplasms including gliomas. The purpose of this review is to discuss the spectrum of intracranial gliomas arising in individuals with NF1 with a focus on recent preclinical and clinical data.
neurofibromatosis
gliomas
glioblastoma
therapeutics
astrocytoma
1. Introduction
Neurofibromatosis type 1 (NF1) is the most common tumor suppressor syndrome, with an incidence of approximately 1 in every 2500 to 3500 births [1]. NF1 is caused by pathogenic variants in the NF1 gene, located at chromosome 17q11.2 [2][3]. Such variants can be familial or occur de novo, with the latter occurring in ~50% of individuals with NF1 [4]. Though NF1 is an autosomal dominant disorder with complete penetrance, there is vast variability in clinical presentation, even in monozygotic twins [5]. There are some genotype-phenotype correlations for specific NF1 variants [6][7], but much of the variability in phenotype has been attributed to stochastic events, environmental factors or modifier genes [8][9][10].
The NF1 gene encodes neurofibromin, a cytoplasmatic 2818 amino acid protein that is widely expressed in the neurons and astrocytes of the central nervous system (CNS), as well as Schwann cells in the peripheral nervous system. Neurofibromin has been shown to control cell growth through two major intracellular pathways. First, neurofibromin negatively regulates the RAS pathway signaling through its action on GTPase-activating protein (GAP), stimulating the conversion of GTP-bound RAS to its GDP-bound form [11]. Increased RAS activity leads to the downstream activity of the MEK-ERK pathway as well as the PI3K-Akt-mTOR pathway (Figure 1) [12]. Neurofibromin has also been shown to positively regulate intracellular levels of cyclic adenosine monophosphate (cAMP), which in turn inhibits cell growth in some cells, including astrocytes [13]. Biallelic inactivation of NF1 gene function is required for tumor formation; i.e., the somatic inactivation of the unaffected NF1 allele is a “second hit,” which leads to absence of neurofibromin within affected cells [14][15].
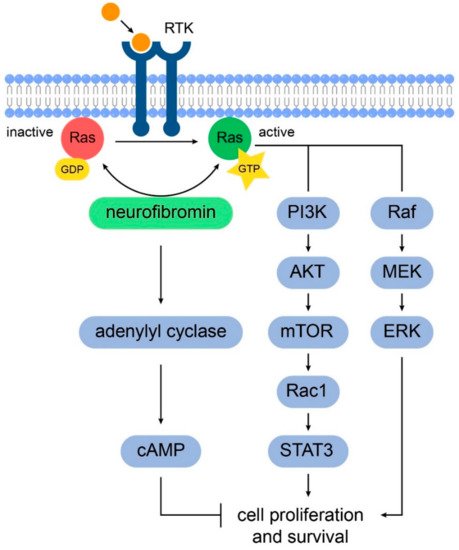
Figure 1. Schematic representation of the signaling pathways involved in NF1 tumorigenesis. Neurofibromin positively regulates adenylyl cyclase to increase intracellular cAMP levels which inhibits glial cell proliferation and survival. Also, neurofibromin promotes the conversion of active GTP-bound RAS to its inactive GDP-bound conformation. In NF1, the increased RAS activity in astrocytes leads to cell proliferation through the downstream activation of the PI3K/AKT/mTOR and RAF-MAK/MEK pathways.
2. Gliomagenesis in Neurofibromatosis Type 1
NF1 is associated with tumors of the peripheral and central nervous system (CNS). The most common CNS tumors in NF1 are gliomas, which are seen in approximately 20% of patients [16][17]. Gliomas usually affect children, with mean age at diagnosis of 4.5 years; the vast majority of such tumors originate within the optic nerves, optic chiasm, and/or hypothalamus. While individuals with NF1 are at higher risk for developing low-grade gliomas compared to high-grade gliomas [18][19], their risk for high-grade glioma is increased by 50-fold when compared to the general population [20][21]. Indeed, high grade gliomas are rare tumors and the reported higher risk in children and adults with NF1 is based on epidemiologic studies and several case series [22].
The World Health Organization (WHO) classification of gliomas has been refined and incorporated molecular parameters, namely 1p/19q codeletion, IDH1/2 mutation, and histone H3-K27M, in addition to histology to define many tumor entities [23]. In general, low-grade gliomas form a group of WHO grade I and grade II tumors while high-grade gliomas form a group of WHO grade III and IV based on malignancy grade, molecular markers and presumed cell of origin. The most common glioma associated with NF1 is pilocytic astrocytoma, a WHO grade I tumor, with the optic pathway glioma being a hallmark lesion [24]. Another low-grade astrocytoma that was reported in children with NF1 is pilomyxoid astrocytoma and the grading was suppressed in the revised 2016 WHO Classification to WHO grade I [25]. In contrast to pilocytic astrocytomas, diffuse astrocytomas, which form WHO grade II, III and IV tumors, are more common in adult individuals with NF with only 12% presenting before the age of 20 [26]. A clinicopathologic study that examined tumors from 100 individuals with NF1 reported pilocytic astrocytoma frequency to be 49% while diffuse astrocytoma to be 27% which included WHO grade II (5%), III (15%), and IV (7%) though this grading used the 2007 WHO Classification [26]. A recently published comprehensive genomic study performed in 23 high grade and 32 low-grade gliomas in individuals with NF1 demonstrated that children developed mostly low-grade gliomas (i.e., 77% of pediatric gliomas were low grade) whereas 78% of tumors in adults were high-grade [27]. The study included whole exome sequencing of tumor and matched blood germline DNA to identify germline and somatic single nucleotide variants, small insertions and deletions, and copy number variants. The NF1 variants observed in germline DNA were typically truncating and led to frameshifts, which did not cluster into specific NF1 protein domains. There was no association between particular patterns of NF1 genetic variants and the risk of developing glioma. The data supported prior reports that a “second-hit” is required to develop tumors [28], as loss of heterozygosity in the NF1 region was detected in the majority of tumors. NF1-associated gliomas were found to have distinct genetic signatures, distinguishing them from those observed in sporadic gliomas, as well as noted to display different genetic landscapes when comparing low- vs. high-grade gliomas. For example, the isocitrate dehydrogenase (IDH) gene mutations (IDH1 and IDH2) are detected in more than 70% of sporadic low-grade gliomas and in the majority of glioblastomas arising from lower grade gliomas [29]. Indeed, individuals with gliomas harboring IDH mutations have better prognosis than those with IDH wild-type [30]. IDH mutations were not detected in gliomas associated with NF1 regardless of grade (Figure 2). This finding may, in part, explain the observation that astrocytomas behave more aggressively than anticipated in adults with NF1 [31]. Another example is that mutations in H3.3 histone genes, frequently found in sporadic pediatric gliomas [32], were absent in all samples regardless of age. Low-grade tumors exhibited fewer mutations that were over-represented in genes of the MAP kinase pathway, while high-grade tumors were characterized by a higher mutation burden and frequent mutations of ATRX, typically co-occurring with alterations of TP53 and cyclin-dependent kinase Inhibitor 2A (CDKN2). DNA methylation assigned NF1-glioma to LGm6, a poorly defined IDH wild-type subgroup enriched with ATRX mutations, which may represent a point of therapeutic intervention, as previous studies have shown that loss of ATRX increases sensitivity to DNA-damaging agents [33][34]. Table 1 summarizes the common molecular differences between NF1-associated gliomas and the LGm6 subgroup of sporadic gliomas [27][35].
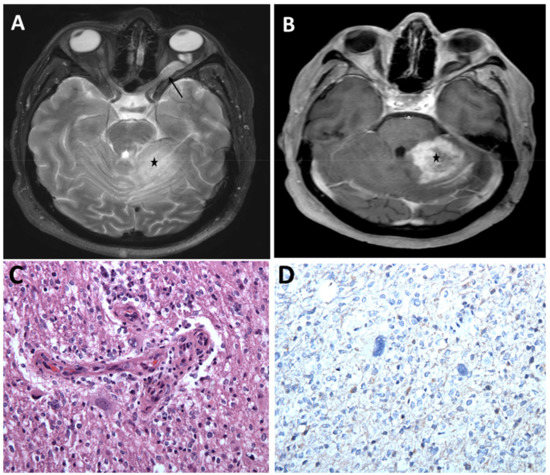
Figure 2. Optic pathway glioma and a high-grade cerebellar glioma in a young adult with NF1. (A) MRI brain, axial T2 sequence showing hyperintense left optic nerve lesion (arrow) and ill-defined hyperintense lesion within the left cerebellum (asterisk) associated with mass effect. (B) Post-contrast T1 sequence showing heterogeneous enhancement of the left cerebellar lesion concerning high grade neoplasm (asterisk). Histopathologic evaluation of the left cerebellar lesion was consistent with glioblastoma, WHO grade IV, IDH wild-type (C) Infiltrating glioma exhibiting atypical cells and vascular endothelial proliferation (H&E, 200×). (D) Tumor cells are negative for IDH1 (R132H) mutant protein (IHC, 200×).
Table 1. Somatic and Germline alterations in NF1-Glioma compared to the LGm6 subgroup of sporadic gliomas.
| Variation | NF1-Glioma | LGm6 Sporadic Glioma | |||
|---|---|---|---|---|---|
| High Grade | Low Grade | High Grade | Low Grade | ||
| Grade IV | Grade III | Grade II | |||
| IDH Wild-Type | 100 | 100 | 100 | 100 | 100 |
| TERT | 47 | 12 | 43 | ||
| ATRX | 38 | 3 | 13 | 42 | 0 |
| CDNK2A | 58 | 19 | 59 | 46 | 17 |
| TP53 | 29 | 0 | 35 | 42 | 0 |
| PTEN | 12 | 0 | 54 | 38 | 0 |
| PIK3CA | 17 | 0 | 13 | 0 | 8 |
| NF1 | 88 | 91 | 22 | 50 | 8 |
| BRAF | 0 | 3 | 4 | 0 | 15 |
| NF1 germline mutation | 92 | 91 | |||
Approximately 50% of low-grade NF1-gliomas displayed an immune signature, T lymphocyte infiltration, and increased neo-antigen load, implying that such tumors may also be targeted via immunotherapies. Such results were confirmed via immunohistochemistry for the T lymphocyte markers CD3 and CD8: the T-cell infiltrates in high-immune NF1-gliomas included cells positive for granzyme B, the cytolytic effector that is upregulated on CD8+ T-cell activation, while B lymphocytes (CD20) and macrophages (CD68) were rare both in high- and low-immune groups [36].
Summary table listing the frequencies (%) of mutations seen in NF1-glioma as studied by D’Angelo, et al. (Nat Med, 2019), separated by high grade (Grade III–IV) and low grade (I–II). This molecular profile most closely correlated to the LGm6 subgroup of pan-glioma cohort from The Cancer Genome Atlas (TCGA) project (Ceccarelli, et al. Cell 2016), which is an IDH-WT group enriched with ATRX mutations.
TERT = TERT copy number variant gain in NF1-Glioma and TERT promotor expression in LGm6 group. ATRX = inactivation of ATRX from any mutation. CDNK2A = loss of copy number variant. TP53 = frameshift or missense mutation in both groups. PTEN = combination of missense and frameshift mutations in the NF-1 glioma group; missense and loss in LGm6 group. PIK3CA = missense and in-frame indel. NF1 = frameshift/truncating. BRAF = missense in NF1-glioma group, missense and frameshift in LGm6 group.
3. Optic Pathway Gliomas
Low-grade gliomas are the most common CNS tumors in the pediatric population, both in children with and without NF1 [37]. Multifocality and predilection for the optic pathways are features commonly associated with low-grade gliomas in NF1. Optic pathway gliomas (OPGs) are the most common brain tumors in individuals with NF1, with the majority classified as pilocytic astrocytoma (PA) [38].
3.1. Genetic and Molecular Pathophysiology
Due to the tumor location, OPG surgery is rarely performed; therefore, there is a relative dearth of genomic and/or microenvironmental studies given the lack of tissue. Multiple studies have been conducted to identify genotype-phenotype associations in NF1-associated OPGs, but reports are conflicting, probably due to the smaller sample sizes [39][40]. Studies have suggested that individuals with mutations in the 5′ tertile (exon 1-21) on NF1 gene have a greater risk of developing OPG, but this was not confirmed in a subsequent study [41][42]. A large cohort study that examined NF1 mutations in 215 NF1 patients (100 of them had OPGs) observed that those with variants in the cysteine/serine-rich domain of the NF1 gene (CSRD, residues 543–909), which is located in 5′ tertile, had higher risk of developing OPGs [39]. A recent genotype-phenotype study reported a more severe phenotype in individuals with NF1 who carry missense mutations affecting one of five neighboring codons 844–848 located outside the GAP-related domain [43]. The study presented 162 individuals heterozygous for a constitutional NF1 missense mutation in one of the five neighboring codons 844–848. The cluster of the recurrent missense mutations reported in this study involving aa 844–848 is located within the CSRD domain, which is likely functionally important, and was originally described by Fahsold et al. [44]. The reported individuals have high prevalence of severe NF1 phenotype, including plexiform and/or spinal neurofibromas, symptomatic OPGs, skeletal abnormalities, and other malignant neoplasms.
Some studies of the tumor microenvironment have highlighted the role of microglia in OPGs, possibly due to the release of monocyte chemoattractant protein-1 (MCP-1) by the gliomas [45][46]. Microglia can have an immunosuppressive role in the tumor microenvironment, through the release of Tumor Growth Factor beta (TGFβ), and Vascular Endothelial Growth Factor (VEGF); findings that raise the possibility of immunomodulation of microglia as a possible therapeutic target in NF1-associated gliomas (Table 2) [47].
Table 2. Clinical trials for NF1 associated gliomas.
| Drug | Target | Tumor | Phase | Age | Endpoints | Status |
|---|---|---|---|---|---|---|
| Vinblastine +/− Bevacizumab NCT02840409 | Cytotoxic/VEGF | LGG | II | 6 months to 18 years | Response rate, OS, PFS, visual outcome measures, OCT | Recruiting |
| Pegylated interferon NCT02343224 | Tumor microenvironment | PA or OPG | II | 3 to 18 years | Response rate | Recruiting |
| Pomalidomide NCT02415153 | Angiogenesis/immunomodulation | NF1-associated CNS tumors | I | 3 to 20 years | Toxicity, MTD | Active, not recruiting |
| Lenalidomide NCT01553149 | Angiogenesis/immunomodulation | PA or OPG | II | 0 to 21 years | Response rate | Active, not recruiting |
| Everolimus (RAD0001) NCT01158651 | mTOR | LGG | II | 1 to 21 years | Response rate | Active, not recruiting |
| Binimetinib (MEK162) NCT02285439 | MEK | LGG | I/II | 1 to 18 years | MTD, response rate | Recruiting |
| Binimetinib (MEK162) NCT01885195 | MEK | Solid tumors with NF1 mutation | II | Older than 18 years | Response rate | Completed (pending results) |
| Selumetinib NCT01089101 | MEK | LGG | I/II | 3 to 21 years | Safety, MTD, Response rate | Recruiting |
| Selumetinib (Selumetinib vs. carboplatin and vincristine) Randomized NCT03871257 | MEK | OPG | III | 2 to 21 years | Event-free survival ∗, visual acuity | Not yet recruiting |
| TAK-580 NCT03429803 | RAF (pan-RAF kinase inhibitor) | LGG | I/II | 1 to 18 years | Toxicity, MTD, 6-month PFS | Recruiting |
Abbreviations: LGG, Low-Grade Glioma; MEK, mitogen activate protein kinase; MTD, maximal tolerated dose; mTOR, mammalian target of rapamycin; OPG, Optic-Pathway Glioma; OS, overall survival; PA, Pilocytic Astrocytoma; PFS, progression free survival; RAF, Rapidly accelerated fibrosarcoma; VEGF, vascular endothelial growth factor. ∗ Event-free survival is the time frame from randomization to the first occurrence of any of the following events: clinical or radiographic disease progression, disease recurrence, second malignant neoplasm, or death from any cause, assessed up to 10 years.
3.2. Clinical Presentation
Children with NF1, 6 years of age or younger, are at the greatest risk for developing OPG, with a slight female preponderance [48][49]. In contrast to non-NF-associated OPGs, NF1-associated OPGs are frequently asymptomatic and some may spontaneously regress [50]. Symptomatic OPGs are almost exclusively diagnosed in children younger than 8 years of age [51], and present with decreased visual acuity, visual field deficits, diminished color perception, optic nerve atrophy, and/or proptosis. Endocrinological problems, especially precocious puberty, may be seen with chiasmatic lesions [52]. It is noteworthy that young children rarely complain of visual loss; hence, reliable and reproducible measures to detect vision changes are necessary. One retrospective study of 54 patients with NF1-associated OPGs demonstrated that 59% had ophthalmological signs at time of presentation [53]. The signs included decreased visual acuity (72%), proptosis (31%) and, in one instance, nystagmus. Precocious puberty was reported in 12 (40%) children with chiasmal OPG, with accelerated linear growth being the first sign [54]. Though there are no current clear prognostic features for OPG progression, patient age, sex, and tumor location may predict disease course and influence treatment initiation. For example, post-chiasmatic tumors lead to vision loss in 62% of patients, compared with 32% in optic nerve and chiasmatic lesions [52]. Age at presentation can be of prognostic value; affected individuals who are younger than 2 years of age or 10 years of age or older at the initial presentation are more likely to have progressive disease that requires treatment [55][56].
3.3. Treatment
While approximately 20% of individuals with NF1 will develop OPGs, only 30–50% of these will be symptomatic and only one-third will require therapeutic intervention [57]. No clear correlation between the imaging features and the biological behavior of these tumors has been found [58]. Hence, close follow up of individuals with NF1-associated OPGs in NF centers using standardized visual assessment metrics is necessary to ensure that children with silent OPGs do not undergo treatment that can lead to unnecessary complications. Common widely available methods that provide objective visual field assessment include Snellen charts, HOTV charts, and Teller Acuity Cards. These methods along with optic disc pallor were evaluated as visual end points in clinical trials for NF1-associated OPGs [59]. Optic coherence tomography (OCT) is being tested to standardize the visual assessment in NF1-associated OPGs [60]. OCT provides an objective assessment of the retinal nerve fiber layer thickness and is a unique noninvasive tool to monitor children with OPG in whom traditional visual assessment is challenging [61]. Another objective noninvasive tool for visual assessment in NF1-associated OPGs is automated tractography of the optic radiations that was validated in a recent study [62]. Screening MRIs for NF1-associated OPGs are not indicated, as the decision to treat is based on clinical, rather than radiographic changes [63], though MRI provides a tool in children with NF1 in whom accurate assessment of visual acuity is not feasible.
In those with declining visual acuity, chemotherapy is considered the mainstay of treatment. First-line chemotherapeutic agents include vincristine and carboplatin [64], while second-line agents include vinblastine [65], vinorelbine [66] and temozolomide [67]. One report showed improvement in visual acuity after using bevacizumab in four cases of refractory OPG (two sporadic and two NF1-associated OPG) [68]. The aim for chemotherapy is to achieve stability and prevent further vision loss, as the currently used chemotherapeutic agents rarely restore premorbid visual acuity [69][70]. As in other tumor suppressor gene syndromes, radiotherapy is usually avoided in NF1-associated OPGs for concern of secondary tumors [71]. Highlighting the risk of radiation in children with NF1, a recent report demonstrated that among NF1-affected individuals with a primary tumor, the risk of secondary neoplasms was 2.8-fold higher in patients who received irradiation [72]. Another risk of radiotherapy in children with NF1 is the development of Moyamoya syndrome due to the radiation exposure to the circle of Willis blood vessels adjacent to the optic pathway [73]. Surgical excision of OPG is not feasible due to the tumor location and is usually reserved for instances of complete loss of vision, severe proptosis, or hydrocephalus [74]. In those with refractory OPGs, small molecule inhibitors have been used in clinical trials. Sorafenib, a multi-kinase inhibitor, is one of the tested agents, but the study was stopped due to unexpected accelerated tumor growth [75]. In this study, 11 patients were evaluated for response and only three had NF1. In vitro studies with sorafenib indicate that this effect is likely related to paradoxical ERK activation. A promising agent is selumetinib, which has shown favorable results in phase II studies of NF1-associated low grade gliomas [76].
References
- Huson, S.M.; Harper, P.S.; Compston, D.A.S. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain 1988, 111, 1355–1381.
- Gutmann, D.H.; Wood, D.L.; Collins, F.S. Identification of the Neurofibromatosis Type 1 Gene Product. Proc. Natl. Acad. Sci. USA 1991, 88, 9658.
- Wallace, M.R.; Marchuk, D.A.; Andersen, L.B.; Letcher, R.; Odeh, H.M.; Saulino, A.M.; Fountain, J.W.; Brereton, A.; Nicholson, J.; Mitchell, A.L.; et al. Type 1 Neurofibromatosis Gene: Identification of a Large Transcript Disrupted in Three NF1 Patients. Science 1990, 249, 181.
- Ars, E.; Kruyer, H.; Morell, M.; Pros, E.; Serra, E.; Ravella, A.; Estivill, X.; Lázaro, C. Recurrent Mutations in the NF1 Gene Are Common among Neurofibromatosis Type 1 Patients. J. Med. Genet. 2003, 40, e82.
- Rieley, M.B.; Stevenson, D.A.; Viskochil, D.H.; Tinkle, B.T.; Martin, L.J.; Schorry, E.K. Variable Expression of Neurofibromatosis 1 in Monozygotic Twins. Am. J. Med. Genet. Part A 2011, 155, 478–485.
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.-A.; Atkin, J.; et al. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients Carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum. Mutat. 2015, 36, 1052–1063.
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 Microdeletions in Neurofibromatosis Type 1: From Genotype to Phenotype. Hum. Mutat. 2010, 31, E1506–E1518.
- Wang, Q.; Montmain, G.; Ruano, E.; Upadhyaya, M.; Dudley, S.; Liskay, R.M.; Thibodeau, S.N.; Puisieux, A. Neurofibromatosis Type 1 Gene as a Mutational Target in a Mismatch Repair-Deficient Cell Type. Hum. Genet. 2003, 112, 117–123.
- Kehrer-Sawatzki, H.; Mautner, V.-F.; Cooper, D.N. Emerging Genotype–Phenotype Relationships in Patients with Large NF1 Deletions. Hum. Genet. 2017, 136, 349–376.
- Easton, D.F.; Ponder, M.A.; Huson, S.M.; Ponder, B.A. An Analysis of Variation in Expression of Neurofibromatosis (NF) Type 1 (NF1): Evidence for Modifying Genes. Am. J. Hum. Genet. 1993, 53, 305–313.
- Basu, T.N.; Gutmann, D.H.; Fletcher, J.A.; Glover, T.W.; Collins, F.S.; Downward, J. Aberrant Regulation of Ras Proteins in Malignant Tumour Cells from Type 1 Neurofibromatosis Patients. Nature 1992, 356, 713–715.
- Ratner, N.; Miller, S.J. A RASopathy Gene Commonly Mutated in Cancer: The Neurofibromatosis Type 1 Tumour Suppressor. Nat. Rev. Cancer 2015, 15, 290–301.
- Hannan, F.; Ho, I.; Tong, J.J.; Zhu, Y.; Nurnberg, P.; Zhong, Y. Effect of Neurofibromatosis Type I Mutations on a Novel Pathway for Adenylyl Cyclase Activation Requiring Neurofibromin and Ras. Hum. Mol. Genet. 2006, 15, 1087–1098.
- Li, Y.; Bollag, G.; Clark, R.; Stevens, J.; Conroy, L.; Fults, D.; Ward, K.; Friedman, E.; Samowitz, W.; Robertson, M.; et al. Somatic Mutations in the Neurofibromatosis 1 Gene in Human Tumors. Cell 1992, 69, 275–281.
- Upadhyaya, M.; Kluwe, L.; Spurlock, G.; Monem, B.; Majounie, E.; Mantripragada, K.; Ruggieri, M.; Chuzhanova, N.; Evans, D.G.; Ferner, R.; et al. Germline and Somatic NF1 Gene Mutation Spectrum in NF1-Associated Malignant Peripheral Nerve Sheath Tumors (MPNSTs). Hum. Mutat. 2008, 29, 74–82.
- Listernick, R.; Charrow, J.; Gutmann, D.H. Intracranial Gliomas in Neurofibromatosis Type 1. Am. J. Med. Genet. 1999, 89, 38–44.
- Listernick, R.; Louis, D.N.; Packer, R.J.; Gutmann, D.H. Optic Pathway Gliomas in Children with Neurofibromatosis 1: Consensus Statement from the Nf1 Optic Pathway Glioma Task Force. Ann. Neurol. 1997, 41, 143–149.
- Albers, A.C.; Gutmann, D.H. Gliomas in Patients with Neurofibromatosis Type 1. Expert Rev. Neurother. 2009, 9, 535–539.
- Korf, B.R. Malignancy in Neurofibromatosis Type 1. Oncologist 2000, 5, 477–485.
- Huttner, A.J.; Kieran, M.W.; Yao, X.; Cruz, L.; Ladner, J.; Quayle, K.; Goumnerova, L.C.; Irons, M.B.; Ullrich, N.J. Clinicopathologic Study of Glioblastoma in Children with Neurofibromatosis Type 1. Pediatr. Blood Cancer 2010, 54, 890–896.
- Rosenfeld, A.; Listernick, R.; Charrow, J.; Goldman, S. Neurofibromatosis Type 1 and High-Grade Tumors of the Central Nervous System. Childs Nerv. Syst. 2010, 26, 663–667.
- Rasmussen, S.A.; Yang, Q.; Friedman, J.M. Mortality in Neurofibromatosis 1: An Analysis Using U.S. Death Certificates. Am. J. Hum. Genet. 2001, 68, 1110–1118.
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820.
- Szudek, J.; Birch, P.; Riccardi, V.M.; Evans, D.G.; Friedman, J.M. Associations of Clinical Features in Neurofibromatosis 1 (NF1). Genet. Epidemiol. 2000, 19, 429–439.
- Helfferich, J.; Nijmeijer, R.; Brouwer, O.F.; Boon, M.; Fock, A.; Hoving, E.W.; Meijer, L.; den Dunnen, W.F.; Bont, E.S. Neurofibromatosis Type 1 Associated Low Grade Gliomas: A Comparison with Sporadic Low Grade Gliomas. Crit. Rev. Oncol. Hematol. 2016, 104, 30–41.
- Rodriguez, F.J.; Perry, A.; Gutmann, D.H.; O’Neill, B.P.; Leonard, J.; Bryant, S.; Giannini, C. Gliomas in Neurofibromatosis Type 1: A Clinicopathologic Study of 100 Patients. J. Neuropathol. Exp. Neurol. 2008, 67, 240–249.
- D’Angelo, F.; Ceccarelli, M.; Tala; Garofano, L.; Zhang, J.; Frattini, V.; Caruso, F.P.; Lewis, G.; Alfaro, K.D.; Bauchet, L.; et al. The Molecular Landscape of Glioma in Patients with Neurofibromatosis 1. Nat. Med. 2019, 25, 176–187.
- Laycock-van Spyk, S.; Thomas, N.; Cooper, D.N.; Upadhyaya, M. Neurofibromatosis Type 1-Associated Tumours: Their Somatic Mutational Spectrum and Pathogenesis. Hum. Genom. 2011, 5, 623–690.
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. New Engl. J. Med. 2009, 360, 765–773.
- Dubbink, H.J.; Taal, W.; van Marion, R.; Kros, J.M.; van Heuvel, I.; Bromberg, J.E.; Zonnenberg, B.A.; Zonnenberg, C.B.; Postma, T.J.; Gijtenbeek, J.M.; et al. IDH1 Mutations in Low-Grade Astrocytomas Predict Survival but Not Response to Temozolomide. Neurology 2009, 73, 1792–1795.
- Guillamo, J.-S.; Créange, A.; Kalifa, C.; Grill, J.; Rodriguez, D.; Doz, F.; Barbarot, S.; Zerah, M.; Sanson, M.; Bastuji-Garin, S.; et al. Prognostic Factors of CNS Tumours in Neurofibromatosis 1 (NF1)A Retrospective Study of 104 Patients. Brain 2003, 126, 152–160.
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver Mutations in Histone H3.3 and Chromatin Remodelling Genes in Paediatric Glioblastoma. Nature 2012, 482, 226–231.
- Koschmann, C.; Calinescu, A.A.; Nunez, F.J.; Mackay, A.; Fazal-Salom, J.; Thomas, D.; Mendez, F.; Kamran, N.; Dzaman, M.; Mulpuri, L.; et al. ATRX Loss Promotes Tumor Growth and Impairs Nonhomologous End Joining DNA Repair in Glioma. Sci. Transl. Med. 2016, 8, 328ra28.
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative Lengthening of Telomeres Renders Cancer Cells Hypersensitive to ATR Inhibitors. Science 2015, 347, 273.
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563.
- Haworth, K.B.; Arnold, M.A.; Pierson, C.R.; Choi, K.; Yeager, N.D.; Ratner, N.; Roberts, R.D.; Finlay, J.L.; Cripe, T.P. Immune Profiling of NF1-Associated Tumors Reveals Histologic Subtype Distinctions and Heterogeneity: Implications for Immunotherapy. Oncotarget 2017, 8, 82037–82048.
- Johnson, K.J.; Cullen, J.; Barnholtz-Sloan, J.S.; Ostrom, Q.T.; Langer, C.E.; Turner, M.C.; McKean-Cowdin, R.; Fisher, J.L.; Lupo, P.J.; Partap, S.; et al. Childhood Brain Tumor Epidemiology: A Brain Tumor Epidemiology Consortium Review. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2716–2736.
- Driever, P.H.; von Hornstein, S.; Pietsch, T.; Kortmann, R.; Warmuth-Metz, M.; Emser, A.; Gnekow, A.K. Natural History and Management of Low-Grade Glioma in NF-1 Children. J. Neuro Oncol. 2010, 100, 199–207.
- Xu, M.; Xiong, H.; Han, Y.; Li, C.; Mai, S.; Huang, Z.; Ai, X.; Guo, Z.; Zeng, F.; Guo, Q. Identification of Mutation Regions on NF1 Responsible for High- and Low-Risk Development of Optic Pathway Glioma in Neurofibromatosis Type I. Front. Genet. 2018, 9.
- Anastasaki, C.; Gao, F.; Gutmann, D.H. Commentary: Identification of Mutation Regions on NF1 Responsible for High- and Low-Risk Development of Optic Pathway Glioma in Neurofibromatosis Type I. Front. Genet. 2019, 10.
- Bolcekova, A.; Nemethova, M.; Zatkova, A.; Hlinkova, K.; Pozgayova, S.; Hlavata, A.; Kadasi, L.; Durovcikova, D.; Gerinec, A.; Husakova, K.; et al. Clusterring of Mutations in the 5′ Tertile of the NF1 Gene in Slovak Patients with Optic Pathway Glioma. Neoplasma 2013, 60.
- Hutter, S.; Piro, R.M.; Waszak, S.M.; Kehrer-Sawatzki, H.; Friedrich, R.E.; Lassaletta, A.; Witt, O.; Korbel, J.O.; Lichter, P.; Schuhmann, M.U.; et al. No Correlation between NF1 Mutation Position and Risk of Optic Pathway Glioma in 77 Unrelated NF1 Patients. Hum. Genet. 2016, 135, 469–475.
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844-848. Am. J. Hum. Genet. 2018, 102, 69–87.
- Fahsold, R.; Hoffmeyer, S.; Mischung, C.; Gille, C.; Ehlers, C.; Kücükceylan, N.; Abdel-Nour, M.; Gewies, A.; Peters, H.; Kaufmann, D.; et al. Minor Lesion Mutational Spectrum of the Entire NF1 Gene Does Not Explain Its High Mutability but Points to a Functional Domain Upstream of the GAP-Related Domain. Am. J. Hum. Genet. 2000, 66, 790–818.
- Watters, J.J.; Schartner, J.M.; Badie, B. Microglia Function in Brain Tumors. J. Neurosci. Res. 2005, 81, 447–455.
- Pong, W.W.; Higer, S.B.; Gianino, S.M.; Emnett, R.J.; Gutmann, D.H. Reduced Microglial CX3CR1 Expression Delays Neurofibromatosis-1 Glioma Formation. Ann. Neurol. 2013, 73, 303–308.
- Karmakar, S.; Reilly, K.M. The Role of the Immune System in Neurofibromatosis Type 1-Associated Nervous System Tumors. CNS Oncol. 2017, 6, 45–60.
- Fisher, M.J.; Loguidice, M.; Gutmann, D.H.; Listernick, R.; Ferner, R.E.; Ullrich, N.J.; Packer, R.J.; Tabori, U.; Hoffman, R.O.; Ardern-Holmes, S.L.; et al. Gender as a Disease Modifier in Neurofibromatosis Type 1 Optic Pathway Glioma. Ann. Neurol. 2014, 75, 799–800.
- Diggs-Andrews, K.A.; Brown, J.A.; Gianino, S.M.; Rubin, J.B.; Wozniak, D.F.; Gutmann, D.H. Sex Is a Major Determinant of Neuronal Dysfunction in Neurofibromatosis Type 1. Ann. Neurol. 2014, 75, 309–316.
- Perilongo, G.; Moras, P.; Carollo, C.; Battistella, A.; Clementi, M.; Laverda, A.; Murgia, A. Spontaneous Partial Regression of Low-Grade Glioma in Children With Neurofibromatosis-1: A Real Possibility. J. Child Neurol. 1999, 14, 352–356.
- Listernick, R.; Charrow, J.; Greenwald, M.; Mets, M. Natural History of Optic Pathway Tumors in Children with Neurofibromatosis Type 1: A Longitudinal Study. J. Pediatr. 1994, 125, 63–66.
- Balcer, L.J.; Liu, G.T.; Heller, G.; Bilaniuk, L.; Volpe, N.J.; Galetta, S.L.; Molloy, P.T.; Phillips, P.C.; Janss, A.J.; Vaughn, S.; et al. Visual Loss in Children with Neurofibromatosis Type 1 and Optic Pathway Gliomas: Relation to Tumor Location by Magnetic Resonance Imaging. Am. J. Ophthalmol. 2001, 131, 442–445.
- Thiagalingam, S.; Flaherty, M.; Billson, F.; North, K. Neurofibromatosis Type 1 and Optic Pathway Gliomas: Follow-up of 54 Patients. Ophthalmology 2004, 111, 568–577.
- Czyzyk, E.; Jóźwiak, S.; Roszkowski, M.; Schwartz, R.A. Optic Pathway Gliomas in Children With and Without Neurofibromatosis 1. J. Child Neurol. 2003, 18, 471–478.
- Listernick, R.; Ferner, R.E.; Piersall, L.; Sharif, S.; Gutmann, D.H.; Charrow, J. Late-Onset Optic Pathway Tumors in Children with Neurofibromatosis 1. Neurology 2004, 63, 1944–1946.
- Campen, C.J.; Gutmann, D.H. Optic Pathway Gliomas in Neurofibromatosis Type 1. J. Child Neurol. 2018, 33, 73–81.
- Fisher, M.J.; Loguidice, M.; Gutmann, D.H.; Listernick, R.; Ferner, R.E.; Ullrich, N.J.; Packer, R.J.; Tabori, U.; Hoffman, R.O.; Ardern-Holmes, S.L.; et al. Visual Outcomes in Children with Neurofibromatosis Type 1–Associated Optic Pathway Glioma Following Chemotherapy: A Multicenter Retrospective Analysis. Neuro-Oncology 2012, 14, 790–797.
- Kornreich, L.; Blaser, S.; Schwarz, M.; Shuper, A.; Vishne, T.H.; Cohen, I.J.; Faingold, R.; Michovitz, S.; Koplewitz, B.; Horev, G. Optic Pathway Glioma: Correlation of Imaging Findings with the Presence of Neurofibromatosis. Am. J. Neuroradiol. 2001, 22, 1963.
- Fisher, M.J.; Avery, R.A.; Allen, J.C.; Ardern-Holmes, S.L.; Bilaniuk, L.T.; Ferner, R.E.; Gutmann, D.H.; Listernick, R.; Martin, S.; Ullrich, N.J.; et al. Functional Outcome Measures for NF1-Associated Optic Pathway Glioma Clinical Trials. Neurology 2013, 81, S15.
- Avery, R.A.; Hwang, E.I.; Ishikawa, H.; Acosta, M.T.; Hutcheson, K.A.; Santos, D.; Zand, D.J.; Kilburn, L.B.; Rosenbaum, K.N.; Rood, B.R.; et al. Handheld Optical Coherence Tomography during Sedation in Young Children with Optic Pathway Gliomas. JAMA Ophthalmol. 2014, 132, 265–271.
- Abdolrahimzadeh, B.; Piraino, D.C.; Albanese, G.; Cruciani, F.; Rahimi, S. Neurofibromatosis: An Update of Ophthalmic Characteristics and Applications of Optical Coherence Tomography. Clin. Ophthalmol. 2016, 10, 851–860.
- De Blank, P.; Fisher, M.J.; Gittleman, H.; Barnholtz-Sloan, J.S.; Badve, C.; Berman, J.I. Validation of an Automated Tractography Method for the Optic Radiations as a Biomarker of Visual Acuity in Neurofibromatosis-Associated Optic Pathway Glioma. Exp. Neurol. 2018, 299, 308–316.
- King, A.; Listernick, R.; Charrow, J.; Piersall, L.; Gutmann, D.H. Optic Pathway Gliomas in Neurofibromatosis Type 1: The Effect of Presenting Symptoms on Outcome. Am. J. Med. Genet. Part A 2003, 122A, 95–99.
- Packer, R.J.; Ater, J.; Allen, J.; Phillips, P.; Geyer, R.; Nicholson, H.S.; Jakacki, R.; Kurczynski, E.; Needle, M.; Finlay, J.; et al. Carboplatin and Vincristine Chemotherapy for Children with Newly Diagnosed Progressive Low-Grade Gliomas. J. Neurosurg. 1997, 86, 747–754.
- Bouffet, E.; Jakacki, R.; Goldman, S.; Hargrave, D.; Hawkins, C.; Shroff, M.; Hukin, J.; Bartels, U.; Foreman, N.; Kellie, S.; et al. Phase II Study of Weekly Vinblastine in Recurrent or Refractory Pediatric Low-Grade Glioma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 1358–1363.
- Cappellano, A.M.; Petrilli, A.S.; da Silva, N.S.; Silva, F.A.; Paiva, P.M.; Cavalheiro, S.; Bouffet, E. Single Agent Vinorelbine in Pediatric Patients with Progressive Optic Pathway Glioma. J. Neuro Oncol. 2015, 121, 405–412.
- Gururangan, S.; Fisher, M.J.; Allen, J.C.; Herndon, J.E.; Quinn, J.A.; Reardon, D.A.; Vredenburgh, J.J.; Desjardins, A.; Phillips, P.C.; Watral, M.A.; et al. Temozolomide in Children with Progressive Low-Grade Glioma. Neuro-Oncology 2007, 9, 161–168.
- Avery, R.A.; Hwang, E.I.; Jakacki, R.I.; Packer, R.J. Marked Recovery of Vision in Children with Optic Pathway Gliomas Treated with Bevacizumab. JAMA Ophthalmol. 2014, 132, 111–114.
- Kalin-Hajdu, E.; Décarie, J.C.; Marzouki, M.; Carret, A.S.; Ospina, L.H. Visual Acuity of Children Treated with Chemotherapy for Optic Pathway Gliomas. Pediatr. Blood Cancer 2014, 61, 223–227.
- Via, P.D.; Opocher, E.; Pinello, M.L.; Calderone, M.; Viscardi, E.; Clementi, M.; Battistella, P.A.; Laverda, A.M.; Da Dalt, L.; Perilongo, G. Visual Outcome of a Cohort of Children with Neurofibromatosis Type 1 and Optic Pathway Glioma Followed by a Pediatric Neuro-Oncology Program. Neuro-Oncology 2007, 9, 430–437.
- Sharif, S.; Ferner, R.; Birch, J.M.; Gillespie, J.E.; Gattamaneni, H.R.; Baser, M.E.; Evans, D.G. Second Primary Tumors in Neurofibromatosis 1 Patients Treated for Optic Glioma: Substantial Risks After Radiotherapy. J. Clin. Oncol. 2006, 24, 2570–2575.
- Bhatia, S.; Chen, Y.; Wong, F.L.; Hageman, L.; Smith, K.; Korf, B.; Cannon, A.; Leidy, D.J.; Paz, A.; Andress, J.E.; et al. Subsequent Neoplasms After a Primary Tumor in Individuals with Neurofibromatosis Type 1. J. Clin. Oncol. 2019.
- Ullrich, N.J.; Robertson, R.; Kinnamon, D.D.; Scott, R.M.; Kieran, M.W.; Turner, C.D.; Chi, S.N.; Goumnerova, L.; Proctor, M.; Tarbell, N.J.; et al. Moyamoya Following Cranial Irradiation for Primary Brain Tumors in Children. Neurology 2007, 68, 932–938.
- Listernick, R.; Ferner, R.E.; Liu, G.T.; Gutmann, D.H. Optic Pathway Gliomas in Neurofibromatosis-1: Controversies and Recommendations. Ann. Neurol. 2007, 61, 189–198.
- Karajannis, M.A.; Legault, G.; Fisher, M.J.; Milla, S.S.; Cohen, K.J.; Wisoff, J.H.; Harter, D.H.; Goldberg, J.D.; Hochman, T.; Merkelson, A.; et al. Phase II Study of Sorafenib in Children with Recurrent or Progressive Low-Grade Astrocytomas. Neuro-Oncology 2014, 16, 1408–1416.
- Fangusaro, J.; Onar-Thomas, A.; Young-Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.; Banerjee, A.; Packer, R.J.; Kilburn, L.B.; Goldman, S.; et al. Selumetinib in Paediatric Patients with BRAF-Aberrant or Neurofibromatosis Type 1-Associated Recurrent, Refractory, or Progressive Low-Grade Glioma: A Multicentre, Phase 2 Trial. Lancet Oncol. 2019.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
905
Revisions:
2 times
(View History)
Update Date:
25 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No