 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anton I. Korbut | + 6303 word(s) | 6303 | 2020-04-30 08:37:57 | | | |
| 2 | Anton I. Korbut | Meta information modification | 6303 | 2020-05-06 20:15:14 | | | | |
| 3 | Rita Xu | Meta information modification | 6303 | 2020-05-09 07:49:51 | | | | |
| 4 | Rita Xu | -594 word(s) | 5709 | 2020-10-29 04:20:33 | | |
Video Upload Options
Glomerular injury is a central feature of diabetic nephropathy. A growing body of evidence indicates a critical role of autophagy in maintaining podocyte integrity and renal function. This process is vital for highly differentiated post-mitotic cells, such as neurons and podocytes. Sodium-glucose cotransporter-2 (SGLT2) inhibitors and dipeptidylpeptidase-4 (DPP4) inhibitors are promising antidiabetic agents introduced into clinical practice in the last decade. The effect of SGLT2 and DPP4 inhibition on glomerular autophagy has not been studied yet. This entry summarizes the study, which have demonstrated, that the SGLT2 inhibitor empagliflozin, the DPP4 inhibitor linagliptin, and a combination of these agents, could reactivate glomerular autophagy in db/db mice, a model of type 2 diabetes. The effect is associated with mitigation of renal hypertrophy, an improvement in glomerular morphology, including a decrease in the severity of podocytopathy, and a slowdown in the growth of albuminuria.
1. Introduction
Diabetic nephropathy is the most common cause for end-stage renal disease worldwide[1]. The pathogenesis of diabetic nephropathy is being studied extensively. A number of studies have identified podocyte injury as a cornerstone in pathogenesis of diabetic kidney disease [2][3]. Podocytes are terminally differentiated cells that wrap the glomerular capillary loops from outside and form slit diaphragms by podocyte foot processes (FPs). Diabetes-induced podocyte injury results in several phenotypic changes, including effacement of FPs, hypertrophy, detachment, death, and/or dedifferentiation. In its turn, the loss or damage of podocytes causes dysfunction of the filtration barrier and increases albuminuria [2].
Some recent studies have indicated an emerging role of autophagy downregulation in diabetic podocytopathy [4][5][6]. Autophagy is a cellular recycling process involving self-degradation and reconstruction of damaged organelles and proteins [6]. The process is vital for highly differentiated post-mitotic cells, such as neurons and podocytes [7]. A growing body of evidence indicates a critical role of autophagy in maintaining podocyte integrity and renal function [6]. Mice with podocyte-specific deletion of autophagic regulators, such as class III PI3K vacuolar protein sorting 34 (Vps34) and the Atg5 gene, develop early proteinuria, progressive glomerulosclerosis, and renal failure [6]. Accordingly, autophagy is considered a potential therapeutic target for renal protection [8][9].
Sodium-glucose cotransporter-2 (SGLT2) inhibitors and dipeptidylpeptidase-4 (DPP4) inhibitors are promising antidiabetic agents introduced into clinical practice in the last decade. The antihyperglycemic effect of SGLT2 inhibitors is mediated by increment of glucosuria, while DPP4 inhibitors realize their activity through an increase in the half-life of incretin hormones. Both SGLT2 and DPP4 inhibitors demonstrated renal protective activity in large-scaled randomized clinical trials. Specifically, in the Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients (EMPA-REG OUTCOME trial) SGLT2 inhibitor empagliflozin reduced the risk of progression to macroalbuminuria, a doubling of the serum creatinine, initiation of renal replacement therapy or death from renal disease in patients with type 2 diabetes [10]. In the Cardiovascular and Renal Microvascular Outcome Study with Linagliptin in Patients with Type 2 Diabetes Mellitus (CARMELINA study), the DPP4 inhibitor linagliptin retarded the albuminuria progression in patients with type 2 diabetes and high cardiovascular risk [11]. It is assumed that double inhibition of SGLT2 and DPP4 may have advantages in terms of renoprotection [12][13].
The mechanisms of protective action of SGLT2 inhibitors and DPP4 inhibitors on diabetic kidney are still a matter for debate. SGLT2 inhibitors have been shown to enhance sodium excretion and suppress glomerulosclerosis and interstitial fibrosis, inflammation and oxidative stress in diabetic kidney [14][15][16]. In addition, DPP4 inhibitors have been showed to reduce glomerular hypertension and hyperfiltration [17][18][19]. Moreover, empagliflozin has been shown to activate autophagy in tubular cells [20][21]. However, the effect of SGLT2 and DPP4 inhibition on glomerular autophagy has not been studied yet [22].
Therefore, we aimed to assess the effect of SGLT2 inhibitor empagliflozin, DPP4 inhibitor linagliptin, and a combination of both agents on glomerular autophagy in a model of type 2 diabetic kidney disease.
2. Data, Model, Applications and Influences
2.1. Methods of the study
The study was carried out at the Center for Genetic Resources of Laboratory Animals at the IC&G SB RAS (Novosibirsk, Russia) with db/db-specific pathogen-free mice (BKS.Cg- Dock7m+/+Leprdb/J, stock #000642) obtained from Jackson Laboratory (Bar Harbor, Maine, USA). The design of the study is presented in Figure 1. After randomization, diabetic db/db mice received empagliflozin (Boehringer Inghelheim, Inghelheim, Germany), linagliptin (Boehringer Inghelheim, Inghelheim, Germany), or the combination of both agents, at a dose of 10 mg/kg of body weight diluted in 200 µl of saline inragastrically. Vehicle-treated mice received only 200 µl inragastrically. All animals were treated once per day for 56 days from 8 to 16 weeks. The control group included the littermate non-diabetic db/+ male mice.
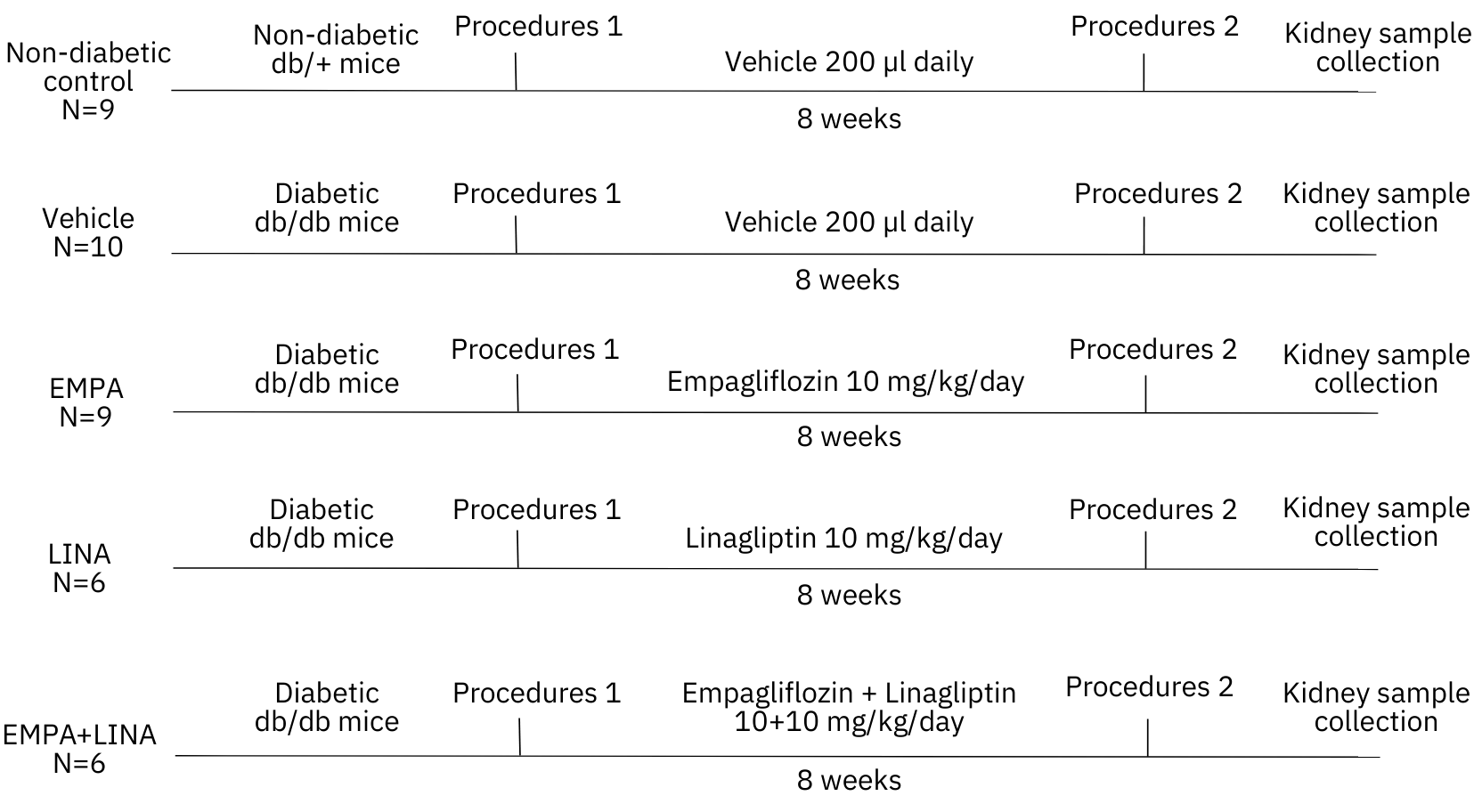
Figure 1. Flowchart of the study. The experiment was started with 8-week-old non-diabetic db+ and diabetic db/db mice. Diabetic mice were randomly assigned for treatment with a vehicle, empagliflozin, linagliptin, or combination of empagliflozin and linagliptin. The total duration of the experiment was 8 weeks. Procedures 1 and 2 included body weight measurement, body composition assessment, blood plasma and urine sampling. EMPA, empagliflozin-treated db/db mice; LINA, linagliptin-treated db/db mice; EMPA + LINA, empagliflozin–linagliptin-treated db/db mice.
Animals were weighed weekly. Body composition was assessed at week 8 and 16 of age by MRI body composition analyzer (Echo Medical Systems, Houston, TX, USA). On the same days, prior to the weighting and body composition study, random blood and urine samples. At week 16, all mice were sacrificed by decapitation under anesthesia. Following laboratory investigations includes plasma levels of glucose, fructosamine, glycated albumin, creatinine, leptin, insulin, ghrelin and PAI-1, urinary albumin-creatinine ratio (UACR).
Kidney samples were obtained for histological assessment, ultrastructural examination, western blot and IHC. To assess renal hypertrophy, kidneys were weighted and adjusted to body weight and lean mass. Mesangial hypertrophy was assessed as volume density (VV) of mesangii in glomeruly. The width of glomerular basement membrane (GBM), mean width and and numerical density (NA) of podocyte FPs was assessed in transmission electron microscopy (TEM) images.
Autophagy and apoptosis regulators in renal cortex were assessed by western blotting using anti-LAMP1, anti-Caspase-3, anti-Bcl-2, anti-LC3B antibody (Abcam, Cambridge, UK, ab25245, ab13847, ab692 and ab48394).
Glomerular autophagy was assessed by with staining for beclin-1 and LAMP-1 (ab62557 and ab25245, respectively, Abcam, Cambridge, UK). The VV of mesangium, glomerular beclin-1, and LAMP-1 positive areas was calculated.
Autophagic compartments (autophagosomes, autolysosomes and lysosomes) in podocytes were defined according to the guidelines for the use and interpretation of assay for monitoring autophagy (3rd edition)[23]. Autophagosomes were identified as compartments with a double membrane, which is separated by an electron-translucent cleft and encircling cytosol and/or organelles appearing morphologically intact [23][24]. Single-membrane, relatively electron-dense organelles, homogenously filled with very tiny granules, were counted as lysosomes (Figure 2a)[25]. Single-membrane compartments with electron dense cytoplasmic material and/or organelles at various stages of degradation were recognized as autolysosomes (Figure 2b) [23][24]. The VV of autophagosomes, autolysosomes and lysosomes in the podocyte cytoplasm was assessed.
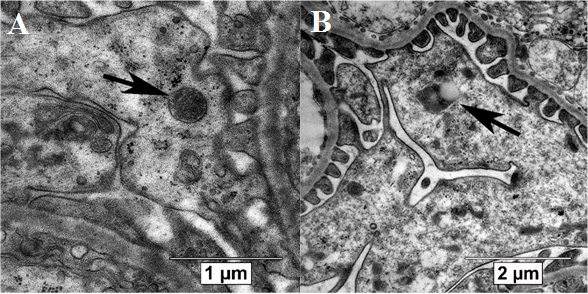
Figure 2. Autophagic compartments in mouse podocytes: lysosomes (black arrow, a) and autolysosome (black arrow, b). TEM, scale bars: 1 μm (a) and 2 µm (b).
2.2. Body Composition and Biochemical Parameters
Diabetic db/db mice had become obese and hyperglycemic prior to the start of experiment. When compared to db/+ controls, 8-week-old db/db mice demonstrated increase in their fat mass and reduction of lean mass and water content (Table 1). In vehicle-treated mice, these changes remained stable throughout the experiment. In actively treated animals, especially in empagliflozin groups, a further increase in the body weight and fat mass was observed.
Table 1. Body weight and body composition in db/+ and db/db mice.
|
Parameter |
Week of age |
db/+ mice (non-diabetic control) |
db/db mice |
|||
|
Vehicle |
EMPA |
LINA |
EMPA+LINA |
|||
|
n |
9 |
10 |
9 |
6 |
6 |
|
|
Body weight, g |
8 |
26.8 (25.0–31.1) |
37.1 (32.1–41.6)*** |
37.4 (28.5–41.5)*** |
36.5 (29.6–40.3)** |
37.3 (35.9–39.5)*** |
|
16 |
24.8 (21.6–27.7)+ |
37.8 (34.3–40.4)*** |
52.6 (42.5–62.2)***##++ |
45.3 (39.5–47.5)***§§+ |
50.4 (46.9–52.3)***#ØØ+ |
|
|
Fat mass, g |
8 |
3.1 (1.9–4.1) |
18.3 (16.2–22.5)*** |
18.4 (13.5–20.6)*** |
18.0 (15.1–19.5)*** |
18.1 (16.7–19.5)*** |
|
16 |
2.7 (2.1–4.5) |
19.6 (17.4–22.7)*** |
28.0 (22.2–35.3)***###++ |
23.4 (20.3–26.4)***##+ |
26.7 (23.2–28.3)***###+ |
|
|
Lean mass, g |
8 |
22.2 (21.0–24.8) |
17.1 (16.3–18.7)*** |
18.2 (13.4–19.8)*** |
16.0 (13.8–17.2)*** |
18.5 (15.8–19.1)*** |
|
16 |
21.2 (19.1–22.5)+ |
16.1 (14.3–22.4)** |
21.4 (19.1–24.4)++ |
17.6 (14.9–20.7)**§+ |
20.6 (19.5–21.5)Ø + |
|
|
Total water, g |
8 |
19.5 (18.4–21.5) |
14.8 (14.5–16.4)*** |
15.8 (11.6–17.6)*** |
12.5 (9.6–14.1)*** |
16.0 (13.7–16.5)*** |
|
16 |
18.4 (16.5–19.4)+ |
14.1 (12.3–19.2)* |
18.8 (16.7–21.4)++ |
15.5 (13.0–18.1)**§+ |
18.2 (17.3–18.7)Ø + |
|
* p < 0.05, ** p < 0.01, *** p < 0.001 vs. non-diabetic control (db/+); # p < 0.05, ## p < 0.01, ### p < 0.001 vs. vehicle-treated db/db mice; § p < 0.05, §§ p < 0.01 vs. empagliflozin group (Mann–Whitney U-test); Ø p < 0.05, ØØ p < 0.01 vs. linagliptin group (Mann–Whitney U-test); + p < 0.05, ++ p < 0.01 vs. with week 8 (Wilcoxon test). EMPA, empagliflozin-treated db/db mice; LINA, linagliptin-treated db/db mice; EMPA+LINA, empagliflozin-linagliptin-treated db/db mice. Data are presented as medians (min – max values).
Diabetic mice demonstrated increased plasma glucose, fructosamine, glycated albumin, leptin, insulin and PAI-1 levels; meanwhile, ghrelin concentrations were decreased significantly (Table 2). Groups of animals were heterogeneous for plasma glucagon levels at week 8 and week 16. Severe hyperglycemia persisted throughout the experiment in vehicle-treated and linagliptin-treated db/db mice.
Table 2. Plasma biochemical parameters and hormones in db/db and db/+ mice.
|
Parameter |
Week of age |
db/+ mice (non-diabetic control) |
db/db mice |
|||
|
Vehicle |
EMPA |
LINA |
EMPA+LINA |
|||
|
n |
9 |
10 |
9 |
6 |
6 |
|
|
Glucose, mmol/l |
8 |
9.5 (5.1–11.4) |
28.5 (16.8–40.1)*** |
23.1 (15.3–35.4)*** |
27.5 (20.6–48.8)*** |
25.9 (20.4–33.0)*** |
|
16 |
9.5 (8.5–12.2) |
32.7 (22.5–53.1) *** |
16.1 (9.9–23.6)***###+ |
29.9 (25.8–34.6)***§§ |
15.2 (11.7–23.5)***ØØ##+ |
|
|
Fructosamine, μmol/l |
8 |
237 (217–249) |
456 (424–511)*** |
480 (425–579)*** |
436 (383–487)*** |
430 (406; 475)*** |
|
16 |
239 (222–296) |
622 (524–672)***++ |
468 (341–491)**+ |
650 (591–691)***§§+ |
380 (342–392)*ØØ+ |
|
|
Glycated albumin, μmol/l |
8 |
107 (103–127) |
227 (206–239)*** |
235 (217–261)*** |
209 (156–256)*** |
217 (203–228)*** |
|
16 |
117 (109–133) |
283 (252–349)***++ |
210 (166–245)**##+ |
315 (225–376)***§§ |
178 (157–182)*###ØØ+ |
|
|
Leptin, ng/ml |
8 |
3.30 (1.40–6.54) |
97.1 (53.2–114.4)*** |
93.4 (80.8–133.1)*** |
92.9 (76.0–118.4)*** |
89.9 (66.6–119.1)*** |
|
16 |
3.80 (1.50–6.30) |
90.0 (21.2–151.4)*** |
136.4 (53.0–171.2)***#+ |
96.5 (51.6–172.6)*** |
139.1 (83.7–200.6)*** |
|
|
Insulin, ng/ml |
8 |
5.59 (3.43–18.0) |
25.6 (11.5–45.2)*** |
21.2 (10.8–40.8)** |
22.7 (15.3–54.0)** |
22.1 (13.8–76.7)*** |
|
16 |
10.0 (2.10–22.2) |
22.1 (9.8–33.2)** |
20.8 (5.95–56.7)* |
13.9 (7.2–23.7) |
33.3 (13.6–72.9)**Ø |
|
|
Glucagon, ng/ml |
8 |
370 (250–2670) |
660 (190–2760) |
450 (290–2180) |
375 (260–2090) |
300 (170–780) |
|
16 |
370 (150–2120) |
605 (260–2960) |
390 (240–860) |
325 (190–810) |
310 (200–460)# |
|
|
Ghrelin, ng/ml |
8 |
0.99 (0.24–4.27) |
0.42 (0.10–1.57)** |
0.50 (0.25–1.15)** |
0.37 (0.25–0.82)** |
0.39 (0.32–1.13)** |
|
16 |
1.47 (0.76–6.31) |
1.27 (0.57–2.26)* |
0.81 (0.28–2.58) |
0.45 (0.20–1.14)***§ |
0.77 (0.08–1.47)* |
|
|
PAI-1, ng/ml |
8 |
1.52 (0.70–3.24) |
2.76 (1.41–4.23)* |
2.07 (1.00–6.01) |
1.84 (0.48–4.20) |
2.25 (1.72–3.91)* |
|
16 |
3.22 (0.40–4.36) |
1.64 (0.09–3.76) |
2.22 (0.79–5.12) |
1.85 (0.63–4.17) |
2.82 (0.98–6.05) |
|
* p < 0.05, ** p < 0.01, *** p < 0.001 vs. non-diabetic control (db/ + ); # p < 0.05, ## p < 0.01, ### p < 0.001 vs. vehicle-treated db/db mice; § p < 0.05, §§ p < 0.01 vs. empagliflozin group (Mann–Whitney U-test) Ø p < 0.05, ØØ p < 0.01 vs. linagliptin group (Mann–Whitney U-test); + p < 0.05, ++ p < 0.01, +++ p < 0.001 vs. week 8 (Wilcoxon test). EMPA, empagliflozin-treated db/db mice; LINA, linagliptin-treated db/db mice; EMPA+LINA, empagliflozin–linagliptin-treated db/db mice. PAI-1, plasminogen activator inhibitor-1. Data are presented as medians (min – max values).
Under the treatment with empagliflozin or empagliflozin–linagliptin combination, db/db mice demonstrated improvement in their glycemic status, which was documented by the reduction in their plasma glucose, fructosamine and glycated albumin (empagliflozin: p = 0.02, p = 0.02, and p = 0.02, respectively; combination: p = 0.03, p = 0.046, and p = 0.046, respectively, vs. week 8). Meanwhile, no significant changes in glycemic control were observed in the linagliptin group. The levels of insulin, glucagon, ghrelin and PAI-1 did not change significantly throughout the experiment in all groups (p > 0.05 week 16 vs. week 8). However, plasma leptin levels were increased significantly in the empagliflozin group and tended to increase in the empagliflozin–linagliptin group (p = 0.03 and p = 0.08, respectively, week 16 vs. week 8). Meanwhile, empagliflozin–linagliptin-treated db/db mice demonstrated lower plasma levels of glucagon as compared with vehicle-treated db/db mice (p = 0.04).
2.3. Albuminuria and Renal Function
A markedly increased albuminuria was detected in all diabetic mice groups at the start of experiment (all p < 0.001, Table 3). Administration of empagliflozin, linagliptin, or both agents decreased albumin excretion (p = 0.007, p = 0.03, and p = 0.04, respectively, vs. week 8). No significant changes in the levels of plasma creatinine were detected throughout the study.
Table 3. Albuminuria and plasma creatinine in db/+ and db/db mice.
|
Parameter |
Week of age |
db/+ mice (non-diabetic control) |
db/db mice |
|||
|
Vehicle |
EMPA |
LINA |
EMPA + LINA |
|||
|
n |
9 |
10 |
9 |
6 |
6 |
|
|
UACR, mg/mmol |
8 |
1.50 (0.60–4.00) |
14.8 (10.0–26.9)*** |
16.3 (12.8–30.0)*** |
26.8 (19.4–54.7)*** |
23.5 (14.3–41.4)*** |
|
16 |
1.80 (0.10–3.10) |
21.4 (14.4–29.4)*** |
11.4 (4.00–22.7)***###++ |
6.9 (4.40–22.4)***##+ |
10.6 (4.50–17.6)***##+ |
|
|
Plasma creatinine, μmol/l |
8 |
67.0 (47.0–78.0) |
76.4 (70.2–84.2) |
70.5 (54.0–82.2) |
70.3 (52.8–103.5) |
70.4 (66.6– 82.2) |
|
16 |
61.8 (54.0–71.7) |
75.6 (63.9–89.4)** |
81.3 (55.8–91.2)* |
71.7 (67.8–99.0)** |
80.4 (65.7–91.2)** |
|
* p < 0.05, ** p < 0.01, *** p < 0.001 vs. non-diabetic control (db/+ mice), Mann–Whitney U-test; ## p < 0.01, ### p < 0.001 vs. vehicle-treated db/db mice; + p < 0.05, ++ p < 0.01 vs. with week 8, Wilcoxon test. EMPA, empagliflozin-treated db/db mice; LINA, linagliptin-treated db/db mice; EMPA+LINA, empagliflozin–linagliptin-treated db/db mice. UACR, urinary albumin-to-creatinine ratio. Data are presented as medians (min - max values).
2.4. Renal Hypertrophy, Glomerular and Podocyte Morphology
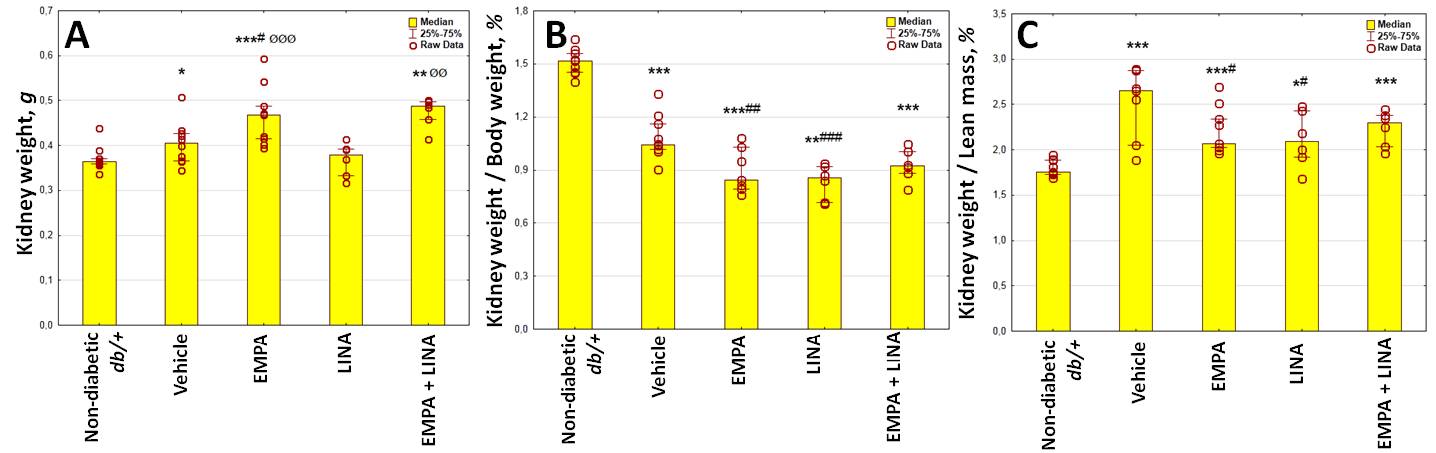
Vehicle-treated db/db mice demonstrated increase in their kidney weight and kidney weight/lean mass ratio, whereas kidney weight/body weight ratio was decreased (p = 0.048, p = 0.0007 and p = 0.00002 vs. non-diabetic db/+ mice, respectively, Figure 3). The kidney weight/body weight and kidney weight/lean mass ratio was lower in empagliflozin and linagliptin-treated mice as compared to vehicle-treated animals (all p < 0.05).
Vehicle-treated db/db mice demonstrated typical morphological signs of diabetic nephropathy, including mesangial expansion, thickening of glomerular basement membrane (GBM), and effacement of podocyte foot processes (FPs). These changes were quantified by morphometric analysis (Table 4, Figure 4).
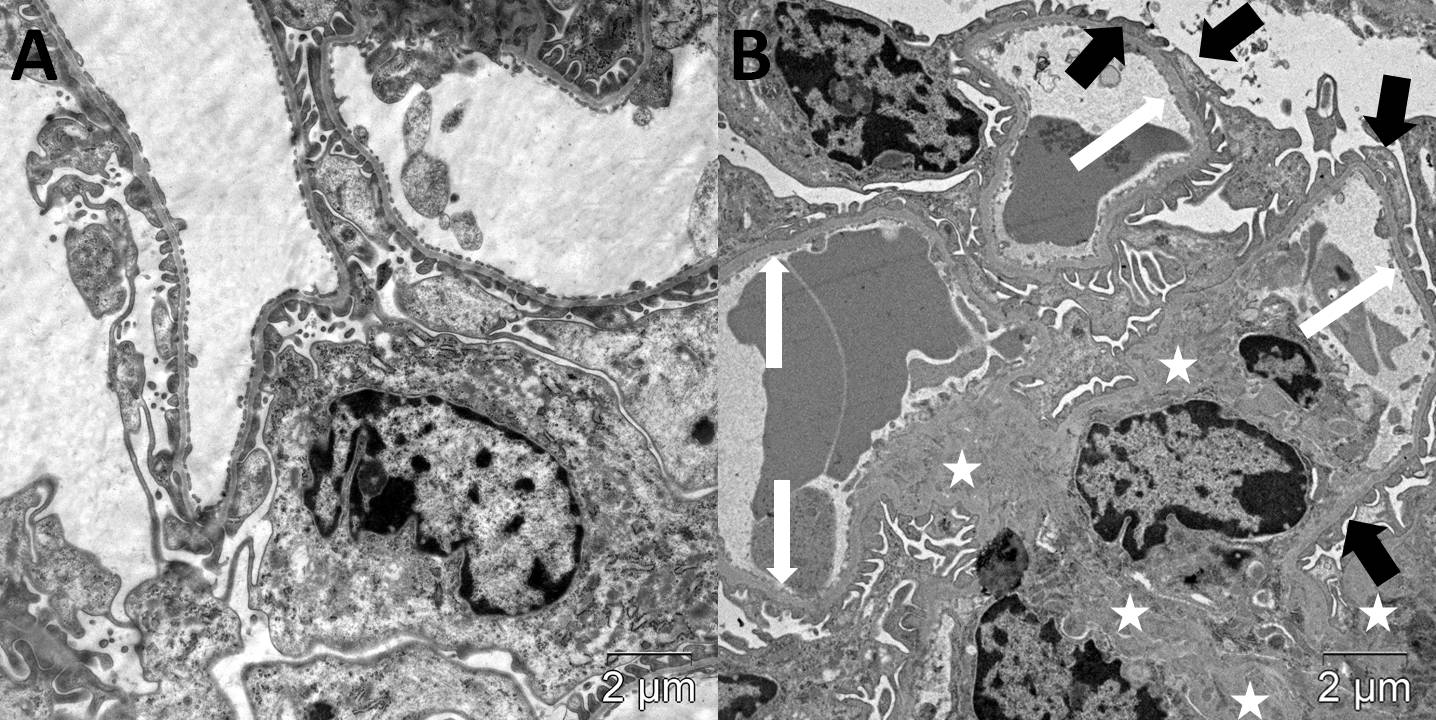
Table 4. Glomerular structural parameters in db/+ and db/db mice.
|
Parameter |
db/+ mice (non-diabetic control) |
db/db mice |
|||
|
Vehicle |
EMPA |
LINA |
EMPA+LINA |
||
|
n |
9 |
10 |
9 |
6 |
6 |
|
Mesangial fractional volume, % |
14.4 |
38.6 |
25.5 |
31.1 |
28.5 |
|
GBM width, nm |
135 |
163 |
139 |
150 |
146 |
|
Width of podocyte FPs, nm |
220 |
372 |
203 |
189 |
202 |
|
NA of podocyte FPs, nm–1 |
3.39 |
2.73 |
3.31 |
3.33 |
3.25 |
* p < 0.05, ** p < 0.01, *** p < 0.001 vs. non-diabetic control (db/ + ); # p < 0.05, ## p < 0.01, ### p < 0.001 vs. vehicle-treated db/db mice; EMPA, empagliflozin-treated db/db mice; LINA, linagliptin-treated db/db mice; EMPA+LINA, empagliflozin–linagliptin-treated db/db mice; GBM, glomerular basement membrane; FPs, foot processes; NA, numerical density. Data are presented as medians (min-max values).
The structural changes in the kidneys were mitigated by empagliflozin, linagliptin, and combined treatment. Specifically, fractional mesangial volume and mean GBM width were reduced (p = 0.0008 and p = 0.02 for empagliflozin; p = 0.002 and p = 0.04 for linagliptin; p = 0.03 and p = 0.04 for combination, respectively). The mean width of podocyte FPs decreased in empagliflozin-treated, linagliptin-treated and empagliflozin–linagliptin-treated mice (p = 0.005, p = 0.02, and p = 0.004, respectively); accordingly, NA of FPs increased in these groups (p = 0.03, p = 0.04, and p = 0.03, respectively).
2.5. Expression of Autophagy and Apoptosis Regulators in the Renal Cortex
Western blotting analysis showed protein expression of LC3B, an autophagosome marker, to be significantly elevated in renal cortex in vehicle-treated db/db mice compared to controls (p=0.008, Figure 5). We found decrease in the protein expression of LC3B in db/db mice treated with empagliflozin and the combination of empagliflozin and linagliptin (both p = 0.008 vs. vehicle group). Protein expression of LAMP-1, a lysosomal marker, was reduced significantly in vehicle-treated db/db mice when compared to non-diabetic db/+ mice (p = 0.008). In the empagliflozin group, the amount of LAMP-1 was restored (p = 0.03 vs. vehicle-treated db/db mice). Although empagliflozin–linagliptin-treated db/db mice demonstrated similar changes in LAMP-1 protein expression, statistical significance was not achieved (p = 0.15 vs. vehicle-treated db/db mice).
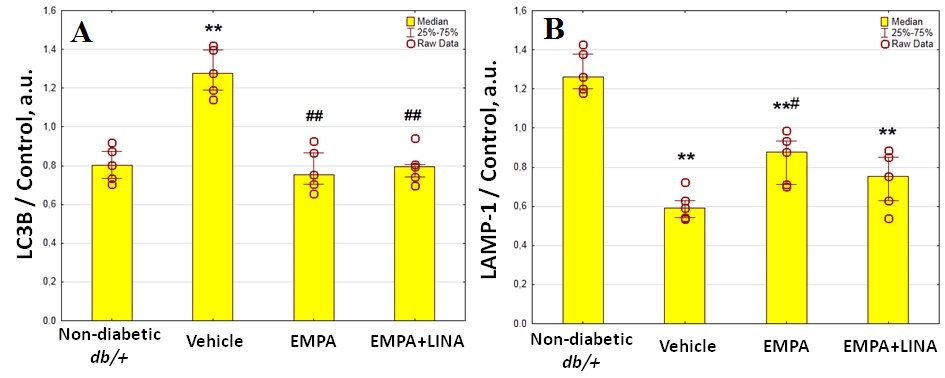

Figure 5. Protein expression of LC3B, an autophagosome marker (a), and LAMP-1, a lysosome marker (b), in the renal cortex from non-diabetic db/+ mice and diabetic db/db mice treated with vehicle, empagliflozin, or empagliflozin–linagliptin; (c) representative immunoblots of LC3B, LAMP-1 and β-actin as the control. Data are presented as a bar graph (median, lower and upper quartile) with individual data set (dots); a.u., arbitrary unit; ** p < 0.01 vs. non-diabetic db/+ mice; # p < 0.05, ## p < 0.01 vs. vehicle-treated db/db mice (Mann–Whitney U-test).
The levels of caspase-3 expression, an apoptosis marker, were elevated markedly in vehicle-treated db/db mice (p = 0.008, Figure 6). The expression of caspase-3 tended to be lower in empagliflozin-treated db/db mice (p = 0.095 vs. vehicle-treated db/db mice); it was significantly lower in db/db mice treated with empagliflozin and linagliptin (p = 0.008 vs. vehicle-treated db/db mice).
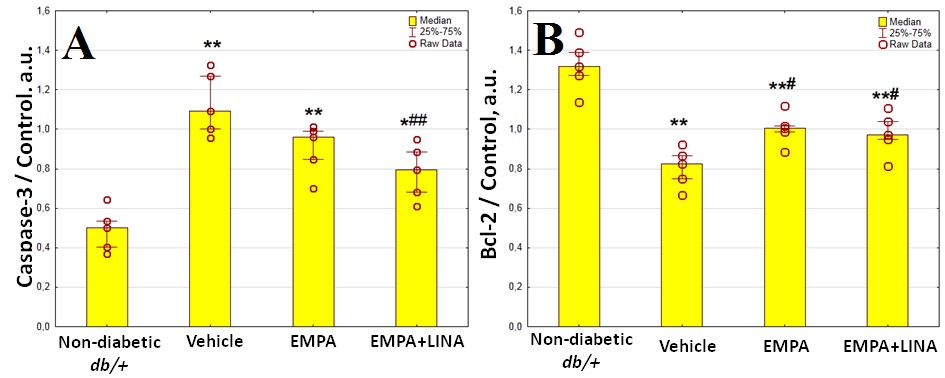
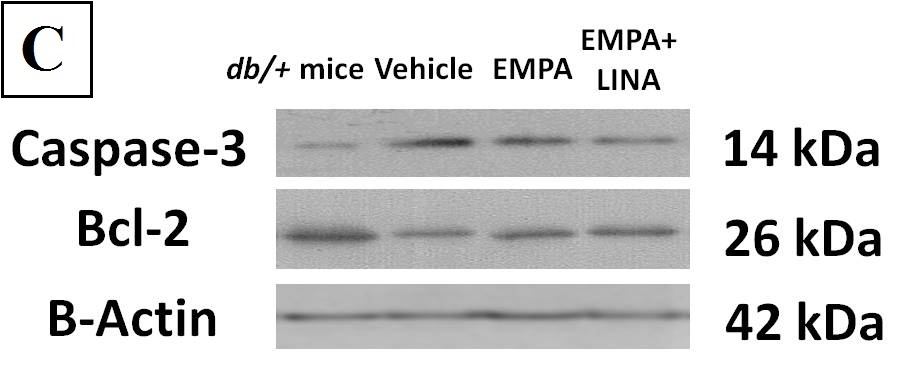
Figure 6. Protein expression of caspase-3, a regulator of apoptosis (a), and Bcl-2, an anti-apoptotic protein (b) in the renal cortex from non-diabetic db/+ mice and diabetic db/db mice treated with vehicle, empagliflozin, and empagliflozin–linagliptin; (c) representative immunoblots of caspase-3, Bcl-2 and β-actin as the control. Data are presented as a bar graph (median, lower and upper quartile) with individual data set (dots); a.u., arbitrary unit; * p < 0.05, ** p < 0.01 vs. non-diabetic db/+ mice; # p < 0.05, ## p < 0.01 vs. vehicle-treated db/db mice (Mann–Whitney U-test).
The expression of Bcl-2, an anti-apoptotic protein, was lower in the kidneys of vehicle-treated db/db mice as compared to db/+ controls (p = 0.008, Figure 6). The db/db mice treated with empagliflozin and empagliflozin–linagliptin demonstrated increase in Bcl-2 expression when compared to vehicle-treated animals (p = 0.02 and p = 0.046, respectively).
2.6. Glomerular Expression of the Autophagy Markers
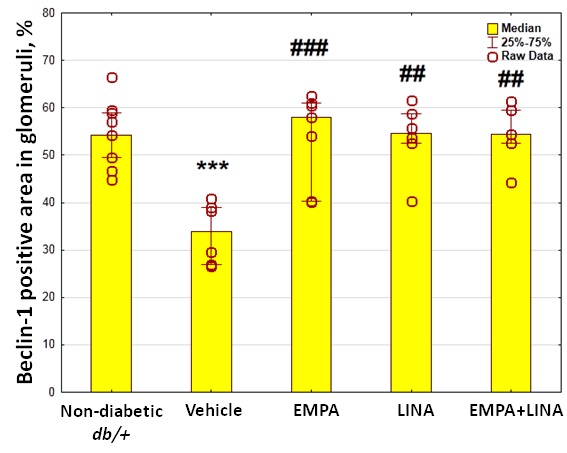
The glomerular expression of beclin-1 and LAMP-1, two markers attributed to autophagy, was assessed by immunohistochemistry (IHC). Vehicle-treated db/db mice demonstrated a reduction of beclin-1-positive areas in glomeruli (p = 0.00002 vs. non-diabetic db/+ mice). This reduction was mitigated by empagliflozin, linagliptin, or empagliflozin–linagliptin treatment (p = 0.0003, p = 0.001 and p = 0.001, respectively vs. vehicle-treated db/db mice, Figure 7).
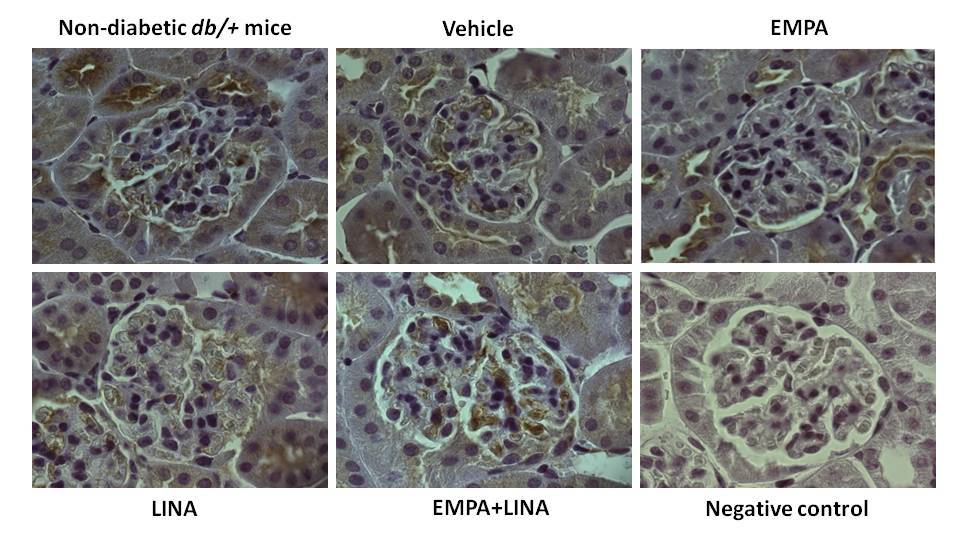
The area of the staining for LAMP-1 was reduced in vehicle-treated db/db mice as compared to non-diabetic db/+ controls (p = 0.01). Administration of empagliflozin, linagliptin or both agents preserved this reduction (p = 0.002, p = 0.04 and p = 0.01 vs. vehicle-treated db/db mice respectively, Figure 8).
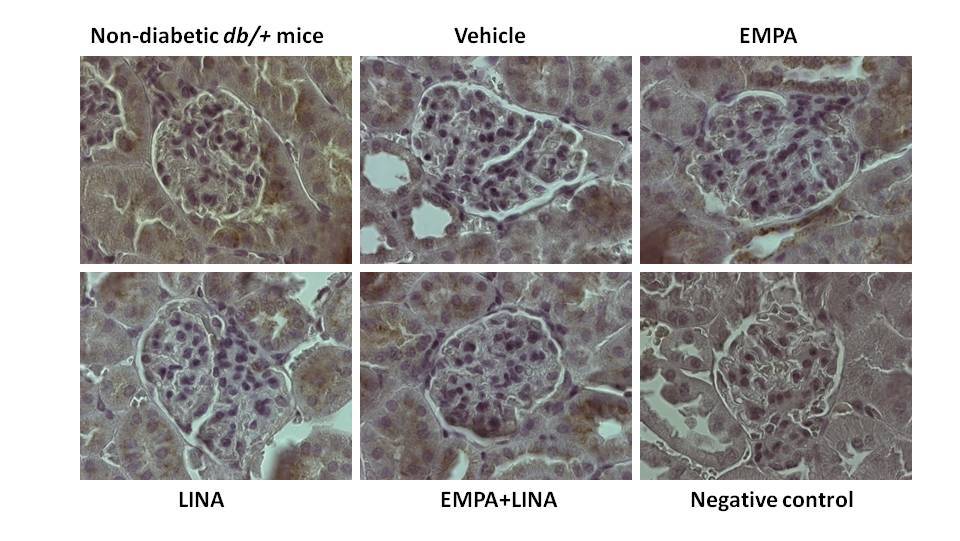
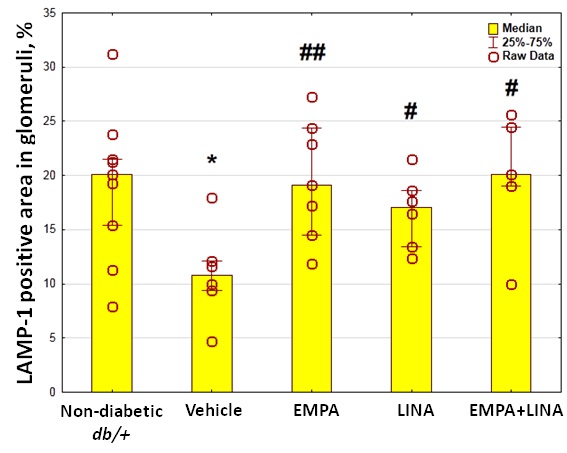
Figure 8. Glomerular LAMP-1 expression in non-diabetic db/+ mice and diabetic db/db mice. Top: IHC-pictures of glomeruli of non-diabetic db/+ mice and db/db mice treated by vehicle, empagliflozin, linagliptin, or empagliflozin–linagliptin; magnification ×400; Bottom: Quantitative analysis of LAMP-1-positive areas in glomeruli. Data are presented as a bar graph (median, lower and upper quartile) with individual data set (dots); * p < 0.05 vs. non-diabetic db/+ mice; # p < 0.05, ## p < 0.01 vs. vehicle-treated db/db mice (Mann–Whitney U-test).
2.7. Autophagy in Podocytes
The VV of autophagosomes, autolysosomes and lysosomes in podocytes was assessed from TEM images. Vehicle-treated diabetic db/db mice had reduced VV of autophagosomes in podocytes (p = 0.00002); administration of empagliflozin, linagliptin, or both agents increased VV of autophagosomes significantly (p = 0.00004, p = 0.0003, and p = 0.0005, respectively, Table 5).
Table 5. The volume density (VV) of autophagosomes, autolysosomes and lysosomes in podocytes in db/+ and db/db mice.
|
Parameter |
db/+ mice (non-diabetic control) |
db/db mice groups |
|||
|
Vehicle |
EMPA |
LINA |
EMPA+LINA |
||
|
n |
5 |
6 |
5 |
5 |
5 |
|
Autophagosomes, VV, % |
2.10 |
0.80 |
2.09 |
2.23 |
2.14 |
|
Autolysosomes, VV, % |
2.37 |
1.09 |
2.25 |
2.37 |
2.22 |
|
Lysosomes, VV, % |
2.94 |
2.22 |
3.16 |
2.64 |
2.89 |
*** p < 0.001 vs. non-diabetic control (db/ + ); # p < 0.05, ## p < 0.01, ### p < 0.001 vs. vehicle-treated db/db mice; EMPA, empagliflozin-treated db/db mice; LINA, linagliptin-treated db/db mice; EMPA + LINA, empagliflozin–linagliptin-treated db/db mice; VV, volume density. Data are presented as medians (min-max values).
There was a reduction in the count of autolysosomes in vehicle-treated db/db mice (p = 0.00002, Table 5). Nevertheless, diabetic db/db mice treated with empagliflozin, linagliptin or the combination of both demonstrated an increase in VV of autolysosomes (p = 0.0003, p = 0.0005, and p = 0.0005, respectively). The changes in the count of lysosomes under these treatments were similar (p = 0.002, p = 0.04, and p = 0.0005, respectively).
2.8. Relationships between Autophagy Intensity, Glomerular Morphology and Biochemical Parameters
In db/+ and db/db mice, both beclin-1 and LAMP-1 correlated positively with VV of autophagosomes and autolysosomes in podocytes (Table 6). Additionally, the area of the staining for LAMP-1 and a protein amount of LAMP-1 demonstrated a correlation with VV of the lysosomes. The VV of autophagosomes and autolysosomes correlated positively with renal LAMP-1 and Bcl-2. However, VV of autophagosomes, autolysosomes and lysosomes correlated negatively with renal LC3B and caspase-3.
Table 6. Relationships between renal autophagy and apoptosis markers and VV of autophagic compartments in podocytes.
|
Parameter |
Autophagosomes, |
Autolysosomes, |
Lysosomes, |
|
Glomerular beclin-1 (IHC) |
r = 0.58 p = 0.0001, n = 26 |
r = 0.48 p = 0.002, n = 26 |
r = 0.25 p > 0.05, n = 26 |
|
Glomerular LAMP-1 (IHC) |
r = 0.46 p = 0.003, n = 26 |
r = 0.50 p = 0.001, n = 26 |
r = 0.42 p = 0.008, n = 26 |
|
Renal LC3B (WB) |
r = –0.57 p = 0.009, n = 20 |
r = –0.69 p = 0.0008, n = 20 |
r = –0.55 p = 0.01, n = 20 |
|
Renal LAMP-1 (WB) |
r = 0.50 p = 0.02, n = 20 |
r = 0.55 p = 0.01, n = 20 |
r = 0.63 p = 0.003, n = 20 |
|
Renal caspase-3 (WB) |
r = –0.35 p > 0.05, n = 20 |
r = –0.56 p = 0.01, n = 20 |
r = –0.44 p > 0.05, n = 20 |
|
Renal Bcl-2 (WB) |
r = 0.54 p = 0.01, n = 20 |
r = 0.57 p = 0.009, n = 20 |
r = 0.63 p = 0.003, n = 20 |
Data are presented as Spearman’s rank correlation coefficients, p-value and number of observations; IHC, immunohistochemistry; VV, volume density; WB, western blot.
Kidney weight/lean mass ratio, considered as an indicator of renal hypertrophy, was found to correlate negatively with VV of autolysosomes and lysosomes in podocytes (Table 7). Mesangial fractional volume correlated negatively with all studied autophagy parameters. A similar pattern of relationships was demonstrated for GBM width. The correlations between metrics of podocyte FPs and autophagy indicators were also revealed.
Table 7. Relationships between glomerular and podocyte autophagy characteristics and other parameters of renal morphology.
|
Parameter |
Glomerular beclin-1, % |
Glomerular LAMP-1, % |
Autophagosomes in podocytes, |
Autolysosomes in podocytes, VV |
Lysosomes in podocytes, VV |
|
Kidneys weight/lean mass ratio |
r = –0.13 p > 0.05, n = 33 |
r = –0.19 p > 0.05, n = 33 |
r = –0.31 p > 0.05, n = 26 |
r = –0.43 p = 0.007, n = 26 |
r = –0.45 p = 0.007, n = 26 |
|
Mesangial fractional volume |
r = –0.39 p = 0.02, n = 40 |
r = –0.37 p = 0.03, n = 40 |
r = –0.36 p = 0.03, n = 40 |
r = –0.50 p = 0.002, n = 40 |
r = –0.32 p = 0.009, n = 40 |
|
Mean width of GBM |
r = –0.41 p = 0.03, n = 26 |
r = –0.40 p = 0.03, n = 26 |
r = –0.54 p = 0.003, n = 26 |
r = –0.45 p = 0.01, n = 26 |
r = –0.26 p > 0.05, n = 26 |
|
Mean width of podocyte FPs |
r = –0.62 p = 0.0004, n = 26 |
r = –0.29 p > 0.05, n = 26 |
r = –0.58 p = 0.0009, n = 26 |
r = –0.49 p = 0.007, n = 26 |
r = –0.41 p = 0.03, n = 26 |
|
NA of podocyte FPs |
r = 0.27 p > 0.05, n = 26 |
r = 0.32 p > 0.05, n = 26 |
r = 0.54 p = 0.003, n = 26 |
r = 0.47 p = 0.01, n = 26 |
r = 0.35 p > 0.05, n = 26 |
Data are presented as Spearman’s rank correlation coefficients; GBM, glomerular basement membrane; FPs, foot processes; NA, numerical density; VV, volume density
Renal cortex expression of caspase-3, LAMP-2 and Bcl-2 was associated with kidney weight/lean mass ratio (Table 8). The mesangial fractional volume and GBM width correlated positively with renal LC3B and caspase-3. Contrary, renal LAMP-1 and Bcl-2 demonstrated negative correlations with mesangial fractional volume and GBM width. Renal LC3B and caspase-3 were associated with the changes in podocyte FPs.
Table 8. Relationships between expression of autophagy and apoptosis regulators in the renal cortex and parameters of renal and glomerular morphology.
|
Parameter |
Renal LC3B |
Renal LAMP-1 |
Renal caspase-3 |
Renal Bcl-2 |
|
Kidney weight/lean mass ratio |
r = 0.40 p > 0.05 |
r = –0.60 p = 0.005 |
r = 0.64 p = 0.002 |
r = –0.64 p = 0.002 |
|
Mesangial fractional volume |
r = 0.45 p = 0.045 |
r = –0.71 p = 0.0004 |
r = 0.67 p = 0.001 |
r = –0.71 p = 0.0004 |
|
Mean width of GBM |
r = 0.67 p = 0.002 |
r = –0.45 p = 0.045 |
r = 0.69 p = 0.002 |
r = –0.53 p = 0.02 |
|
Mean width of podocyte FPs |
r = 0.56 p = 0.02 |
r = –0.38 p > 0.05 |
r = 0.52 p = 0.02 |
r = –0.44 p = 0.048 |
|
NA of podocyte FPs |
r = –0.46 p = 0.04 |
r = 0.38 p > 0.05 |
r = –0.62 p = 0.004 |
r = 0.46 p = 0.04 |
Data are presented as Spearman’s rank correlation coefficients. n = 20. GBM, glomerular basement membrane; FPs, foot processes; NA, numerical density; VV, volume density
There were negative correlations between indicators of autophagy and glycemic control parameters. Specifically, plasma fructosamine and glycated albumin correlated with renal LAMP-1 (r = −0.65, p = 0.002 and r = −0.42, p = 0.007 respectively), glomerular beclin−1 (r = −0.34, p = 0.04 for both) and LAMP−1 (r = −0.41, p = 0.008; r = −0.42, p = 0.007), podocyte VV of autophagosomes (r = −0.34, p = 0.03; r = −0.32, p = 0.046), autolysosomes (r = −0.34, p = 0.03; r = −0.31, p > 0.05) and lysosomes (r = −0.36, p = 0.02; r = −0.33, p = 0.04). No correlations were found with plasma leptin, insulin, ghrelin and PAI-1. However, plasma glucagon concentrations showed negative correlations with glomerular LAMP-1 (r = −0.39, p = 0.03), VV of autophagosomes (r = −0.44, p = 0.005) and lysosomes (r = −0.38, p = 0.02).
Renal caspase-3 demonstrated strong positive correlations with fructosamine and glycated albumin levels (r = 0.91, p < 0.0001 and r = 0.89, p < 0.0001, respectively); renal Bcl-2 correlated with these parameters negatively (r = −0.66, p = 0.001; r = −0.67, p = 0.001). We found no correlations between autophagy and apoptosis parameters and plasma creatinine levels. Meanwhile, UACR correlated negatively with renal LAMP-1 (r = −0.76, p = 0.001), Bcl-2 (r = −0.81, p < 0.0001), glomerular beclin-1 (r = −0.44, p = 0.006) and VV of autophagosomes (r = −0.47, p = 0.002), autolysosomes (r = −0.48, p = 0.002) and lysosomes (r = −0.53, p = 0.0004) in podocytes. There were positive correlations between UACR and renal LC3B (r = 0.50, p = 0.03) and caspase-3 expression (r = 0.66, p = 0.002).
2.9. Suppression of Renal Autophagy in Diabetes: Markers and Mechanisms
The suppression of macroautophagy in db/db diabetic mice was documented by the reduction of glomerular IHC-staining for beclin-1 and LAMP-1, two autophagy markers, and decrease in the volume density of the autophagic compartments (autophagosomes, autolysosomes, and lysosomes) in the podocytes. We have also revealed the reduced expression of LAMP-1 in the renal cortex by western blotting. The results are in agreement with the data from other researches recorded downregulation of autophagy in diabetic kidneys [4][26][27][28][29].
The data on the enhanced expression of LC3B in the renal cortex, obtained by western blot, at first glance are out of the picture. The LC3 molecule is commonly considered a marker of autophagosomes, although it could also be detected in phagophores [23]. LC3-II is generated by the conjugation of cytosolic LC3-I to phosphatidylethanolamine on the surface of nascent autophagosomes. Accordingly, LC3-II is relatively specifically associated with autophagosomes and autolysosomes [30]. In previous studies, both a decrease in LC3-II/LC3-I ratio [31][32] and an increase in LC3-I/II expression [33] in the kidneys of db/db mice was reported. In podocyte cell culture, the expression of LC3-II was increased under high glucose condition [34]. Similarly, an increased LC3B/LC3A ratio was found in high-fat-diet-induced, obesity-related glomerulopathy in mice [35]. Increased LC3-II expression was previously described in tubular epithelial cells in diabetes [36]. Accumulation of LC3-II could be observed when the fusion of autophagosomes with lysosomes was interrupted by chemical agents [23]. It was demonstrated that endogenous LC3-positive puncta become larger in cells where autophagosome formation is abrogated and are prominent even when LC3-II is not formed. This occurs even with transient and incomplete inhibition of autophagosome biogenesis. This phenomenon is due to LC3-I sequestration to p62 aggregates, which accumulate when autophagy is impaired [30]. In this study, we found negative correlations between LC3B levels in the renal cortex and the volume density of lysosomes and autolysosomes in the podocytes. Taking into account a decrease in VV of autophagosomes and lysosomes, as well as a decrease in the renal expression of other markers of autophagy, we can assume that an increase in LC3B protein could be attributed to compromised autophagy in diabetic kidney.
The mechanisms of autophagy suppression in diabetic kidneys remain a matter of debate. Hyperglycemia was considered an autophagy suppressor in the studies with cultured podocytes [27][37], glomerular endothelial cells [38] and mesangial cells [39]. Kidney macroautophagy is regulated by mammalian target of rapamycin (mTOR) [8][40][41], AMP-activated protein kinase (AMPK)[8], SIRT1[8], Wnt/β-catenin [42], and TGF-β [43] signaling pathways. The downregulation of glomerular autophagy under hyperglycemic condition is considered a result of activation of mTOR pathway [8][44] or suppression of AMPK activity and SIRT1 signaling [39][45][46]. In our study, glomerular autophagy was related negatively to parameters of glycemic status, fructosamine and glycated albumin. As it has been demonstrated previously, advanced glycation end-products impair autophagic flux in the podocytes via mTOR activation and inhibition of the nuclear translocation and activity of the pro-autophagic transcription factor EB (TFEB) [47]. In cultured mesangial cells, advanced glycation end-products inhibited autophagy via the RAGE/STAT5 axis [48]. Therefore, hyperglycemia and related activation of non-enzymatic glycation could be a cornerstone in glomerular autophagy suppression.
In this study, we used leptin receptor-defective db/db mice as a model of type 2 diabetes. In these mice, we found extremely high levels of leptin, which can be explained by obesity and defective leptin signaling. The recent data indicate that leptin plays an important role in the neuroendocrine control of autophagy. It was demonstrated that intravenous or intraperitoneal administration of recombinant leptin induced autophagy in mouse peripheral tissues, including skeletal muscle, heart and liver. Moreover, leptin stimulated canonical autophagy in cultured human or mouse cell lines, a phenomenon that was coupled to the activation of AMPK, as well as the inhibition of mTOR, and this was confirmed by autophagic flux measurements [49]. Thereby, it can be assumed that the defect of leptin receptor is another factor contributing to the suppression of autophagy in db/db mice.
The recorded high insulin levels in plasma and severe hyperglycemia in db/db mice undoubtedly indicate insulin resistance, which is consistent with the literature [50]. Intrinsic interactions between insulin signaling and podocyte autophagy have been proposed. The recent findings suggested that downregulation of podocyte autophagy, observed after long-term exposure to high glucose, results from suppressed sensitivity to insulin [51]. Podocytes express all the elements of the insulin-signaling cascade, such as functional insulin receptor substrate-1 and insulin receptor, and are capable of increasing their glucose uptake when they are stimulated with insulin through glucose transporters, primarily glucose transporter type 4 (GLUT4) [52]. Glomeruli from mice with podocyte-specific knockout of GLUT4 are protected from diabetes-induced hypertrophy, mesangial expansion and albuminuria, and fail to activate the mTOR pathway [53]. The latter is activated in podocytes in diabetic conditions that decrease autophagy activity [44]. Interestingly, rapamycin-induced autophagy increased the glucose uptake in the podocytes and phosphorylated insulin receptor, which caused an increase in insulin sensitivity [27].
In addition to insulin, the activity of autophagy in diabetes can be modified by other hormonal regulators. We recorded a significant decrease in concentrations of ghrelin in the plasma of db/db mice. Ghrelin is a proven enhancer of autophagy, mediating its effect through the activation of AMPK [54]. Additionally, PAI-1 is considered an autophagy activator in immune cells [55]. In a model of vinyl-chloride-induced renal fibrosis, activation of autophagy corresponded to increased PAI-1 expression, while expression of PAI-1 gene in HK-2 cell culture was increased under beclin-1 siRNA exposure [56]. Nevertheless, in our study we failed to find any relationship between plasma PAI-1 levels and autophagy parameters.
Glucagon is shown to regulate autophagy by increasing the number and fragility of lysosomes and apoptotic vacuoles. These effects were primary described in liver and suggested to be organ-specific [57]. In our study, plasma glucagon levels correlated negatively with glomerular LAMP-1 and volume density of autophagosomes and lysosomes in the podocytes. These results do not prove causality; the role of glucagon in regulation of renal autophagy in diabetes requires further research.
2.10. Renal Autophagy Suppression in Pathophysiology of Diabetic Kidney Disease
In our study, the suppression of autophagy was related to severity of the classical signs of diabetic nephropathy, including mesangial expansion, GBM thickening and podocytopathy. It was suggested that the presence or activation of autophagy could play a protective role in human podocytes against high glucose-induced insulin resistance and cell injury [27]. In its turn, downregulation of autophagy is critical for podocyte survival in hyperglycemic conditions [51]. In the study with autophagy-deficient models of diabetes, blocking of autophagy in podocytes or glomerular endothelial cells caused podocytopathy and leaky glomerular filtration barriers [38]. The deficiency of autophagy activation in KkAy diabetic mice resulted in more severe proteinuria and impaired renal function [29]. In agreement with these data, we found an inverse association between morphometric parameters of glomerular autophagy, podocytopathy and albuminuria in the model of type 2 diabetes.
Apoptosis is considered another contributor to podocyte depletion in diabetes [44]. In this study, we investigated the expression of caspase-3, an apoptosis regulator, and Bcl-2, an anti-apoptotic protein, in the renal cortex of db/db mice. We found increased protein expression of caspase-3; meanwhile, Bcl-2 expression was diminished. Thus, we recorded reciprocal changes in the levels of autophagy and apoptosis markers. It was demonstrated recently that autophagy can regulate apoptosis following molecular interactions between the key proteins of these processes, including members of Bcl-2 family, autophagy proteins, and caspases [58]. Firstly, beclin-1 can bind to Bcl-2, which results in inhibition of autophagy [58]. Contrary, release of beclin-1 from the complex with Bcl-2 and phosphorylation of beclin-1 or Bcl-2 promote autophagy [23]. In addition, cleavage of beclin-1 by caspase-3 inactivates autophagy and promotes apoptosis [59]. This could be an explanation for the concomitant increase in caspase-3 and decrease in beclin-1 expression recorded in our study.
2.11. Reactivation of Autophagy: a Missing Link in the Mechanism of Renoprotection under SGLT2 and DPP4 Inhibition?
In this work, we demonstrate that empagliflozin and linagliptin, both as monotherapy and in combination, reactivate glomerular autophagy in db/db diabetic mice. In the previous studies, empagliflozin restored autophagy in the tubular cells in a model of streptozotocin-induced diabetes [20], and in the heart in a model of myocardial infarction and type 2 diabetes [60]. Canagliflozin, another SGLT2 inhibitor, activated autophagy in immune cells [31]. A negative correlation between beclin-1 and DPP4 activity was observed in patients with coronary heart disease [61]. The DPP4 inhibitors vildagliptin and sitagliptin improved cardiac function after myocardial infarction through activation of autophagy in the rodent models of diabetes [62][63]. Sitagliptin activated autophagy in cardiac muscle in Zucker diabetic fatty rats, in high-glucose cultured embryonic heart myoblastic (H9c2) cells [61] and in the liver of ob/ob mice, a mouse model of genetic obesity and diabetes [64].
According to our data, empagliflozin, both alone and in combination with linagliptin, contributed to preventing changes in the renal expression of apoptosis regulators, caspase-3 and Bcl-2. In the previous studies, empagliflozin have demonstrated decreased expression of pro-apoptotic proteins in cell cultures of human renal proximal tubular cells (hRPTCs) [20] and in the kidney in streptozotocin-induced diabetic rats [22]. Taking into account the close relationships in the regulation of autophagy and apoptosis, normalization of the expression of apoptosis regulators could also contribute to the reactivation of autophagy under empagliflozin treatment.
It is well known that both SGLT2 and DPP4 inhibitors generate glucose-dependent and glucose-independent effects in the targeted organs [14][15][16][17][18][19]. In our study, autophagy was reactivated by both empagliflozin and linagliptin, while glucose levels were reduced, but not to the normal range, only under empagliflozin administration. We did not observe antihyperglycemic effects of lingagliptin in db/db mice in our study, although we proved renal benefits of the agent. Similarly, in previous studies, linagliptin ameliorated glomerular changes in db/db mice, despite of the lack of antihyperglycemic activity [65][66]. This suggests a glucose-independent mechanism of autophagy reactivation. It was demonstrated that vildagliptin suppressed Beclin-1–Bcl-2 interaction and increased both LC3-II protein level and autophagosomes in the diabetic heart [62]. In the liver of ob/ob mice, sitagliptin increased the levels of phosphorylation of AMPK and decreased those of mTOR [64]. The activation of the AMPK signaling pathway in immune cells was proven for canagliflozin [31]. Further identification of the molecular mechanisms for autophagy activation under SGLT2 and DPP4 inhibition is a challenge for future research.
3. Conclusions
SGLT2 inhibitors and DPP4 inhibitors are relatively new classes of antihyperglycemic agents with distinct renoprotective actions. Identifying the renal effects of these drugs is a challenge to modern medicine. In our study, we demonstrated that both empagliflozin, a SGLT2 inhibitor, and linagliptin, a DPP4 inhibitor, attenuate diabetic kidney disease by promoting glomerular autophagy in db/db mice, a model of type 2 diabetes. The acceleration of autophagy flux under empagliflozin and linagliptin treatment is associated with mitigation of glomerular sclerosis, preservation of podocyte morphology, as well as with a slowdown in the growth of albuminuria. Upregulation of beclin-1 and downregulation of caspase-3 could be suggested as possible pathways in autophagy promotion. The data provide a new explanation for the mechanism of the renoprotective action of SGLT2 inhibitors and DPP4 inhibitors in diabetic kidney disease.
4. Abbreviations
|
AMPK |
AMP-activated protein kinase |
|
DPP4 |
Dipeptidylpeptidase-4 |
|
GLUT4 |
Gucose transporter type 4 |
|
IHC |
Immunohistochemistry |
|
mTOR |
mammalian Target of rapamycin |
|
NA |
Numerical density |
|
LAMP-1 |
Lysosomal-associated membrane protein-1 |
|
PAI-1 |
Plasminogen activator inhibitor-1 |
|
PFs |
Foot processes |
|
SGLT2 |
Sodium-glucose cotransporter-2 |
|
TEM |
Transmission electron microscopy |
|
UACR |
Urinary albumin-to-creatinine ratio |
|
VV |
Volume density |
References
- Angela C Webster; Evi V Nagler; Rachael L Morton; Philip Masson; Chronic Kidney Disease. The Lancet 2017, 389, 1238-1252, 10.1016/s0140-6736(16)32064-5.
- Jamie Lin; Katalin Susztak; Podocytes: the Weakest Link in Diabetic Kidney Disease?. Current Diabetes Reports 2016, 16, 45-45, 10.1007/s11892-016-0735-5.
- Silvia Maestroni; Gianpaolo Zerbini; Glomerular endothelial cells versus podocytes as the cellular target in diabetic nephropathy. Acta Diabetologica Latina 2018, 55, 1105-1111, 10.1007/s00592-018-1211-2.
- Mako Yasuda-Yamahara; Shinji Kume; Atsuko Tagawa; Hiroshi Maegawa; Takashi Uzu; Emerging role of podocyte autophagy in the progression of diabetic nephropathy.. Autophagy 2015, 11, 2385-6, 10.1080/15548627.2015.1115173.
- Na Liu; Liuqing Xu; Yingfeng Shi; Shougang Zhuang; Podocyte Autophagy: A Potential Therapeutic Target to Prevent the Progression of Diabetic Nephropathy. Journal of Diabetes Research 2017, 2017, 1-6, 10.1155/2017/3560238.
- Tien An Lin; Victor Chien-Chia Wu; Chao-Yung Wang; Autophagy in Chronic Kidney Diseases. Cells 2019, 8, 61, 10.3390/cells8010061.
- Michio Nagata; Podocyte injury and its consequences. Kidney International 2016, 89, 1221-1230, 10.1016/j.kint.2016.01.012.
- Danyi Yang; Man J. Livingston; Zhiwen Liu; Guie Dong; Ming Zhang; Jian-Kang Chen; Zheng Dong; Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential. Cellular and Molecular Life Sciences 2017, 75, 669-688, 10.1007/s00018-017-2639-1.
- Yingmei Zhang; Adam Whaley-Connell; James R. Sowers; Jun Ren; Autophagy as an emerging target in cardiorenal metabolic disease: From pathophysiology to management. Pharmacology & Therapeutics 2018, 191, 1-22, 10.1016/j.pharmthera.2018.06.004.
- Christoph Wanner; Silvio E. Inzucchi; Bernard Zinman; Masarori Wakisaka; Anil Pareek; Nitin Chandurkar; Kumar Naidu; Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes.. New England Journal of Medicine 2016, 375, 1801-2, 10.1056/NEJMc1611290.
- Julio Rosenstock; Vlado Perkovic; Odd Erik Johansen; Mark E. Cooper; Steven E. Kahn; Nikolaus Marx; John H. Alexander; Michael Pencina; Robert D. Toto; Christoph Wanner; et al.Bernard ZinmanHans Juergen WoerleDavid BaanstraEgon PfarrSven SchnaidtThomas MeinickeJyothis T. GeorgeMaximilian Von EynattenDarren K. McGuire Effect of Linagliptin vs Placebo on Major Cardiovascular Events in Adults With Type 2 Diabetes and High Cardiovascular and Renal Risk. JAMA 2018, 321, 69-79, 10.1001/jama.2018.18269.
- Yochai Birnbaum; Mandeep Bajaj; Hsiu-Chiung Yang; Yumei Ye; Combined SGLT2 and DPP4 Inhibition Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Nephropathy in Mice with Type 2 Diabetes. Cardiovascular Drugs and Therapy 2018, 32, 135-145, 10.1007/s10557-018-6778-x.
- Honghong Zou; Baoqin Zhou; Gaosi Xu; SGLT2 inhibitors: a novel choice for the combination therapy in diabetic kidney disease.. Cardiovascular Diabetology 2017, 16, 65, 10.1186/s12933-017-0547-1.
- Paola Fioretto; Alberto Zambon; Marco Rossato; Luca Busetto; Roberto Vettor; SGLT2 Inhibitors and the Diabetic Kidney. Diabetes Care 2016, 39, S165-S171, 10.2337/dcs15-3006.
- Anton I. Korbut; Vadim V. Klimontov; Empagliflozin: a new strategy for nephroprotection in diabetes. Diabetes mellitus 2017, 20, 75-84, 10.14341/dm8005.
- Josselin Nespoux; Volker Vallon; SGLT2 inhibition and kidney protection.. Clinical Science 2018, 132, 1329-1339, 10.1042/CS20171298.
- Anton Ivanovich Korbut; Vadim V. Klimontov; Incretin-based therapy: renal effects. Diabetes mellitus 2016, 19, 53-63, 10.14341/dm7727.
- A.J. Scheen; Pierre Delanaye; Renal outcomes with dipeptidyl peptidase-4 inhibitors. Diabetes & Metabolism 2018, 44, 101-111, 10.1016/j.diabet.2017.07.011.
- Shreyasi Gupta; Utpal Sen; More than just an enzyme: Dipeptidyl peptidase-4 (DPP-4) and its association with diabetic kidney remodelling. Pharmacological Research 2019, 147, 104391, 10.1016/j.phrs.2019.104391.
- Yu Ho Lee; Sang Hoon Kim; Jun Mo Kang; Jin Hyung Heo; Ng-Jin Kim; Seon Hwa Park; Minji Sung; Jaehee Kim; Jisu Oh; Dong Ho Yang; Sang-Ho Lee; So-Young Lee; Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy.. American Journal of Physiology-Renal Physiology 2019, 317, F767-F780, 10.1152/ajprenal.00565.2018.
- Chih-Chao Yang; Yen-Ta Chen; Christopher Glenn Wallace; Kuan-Hung Chen; Ben-Chung Cheng; Pei-Hsun Sung; Yi-Chen Li; Sheung-Fat Ko; Hsueh-Wen Chang; Hon-Kan Yip; Early administration of empagliflozin preserved heart function in cardiorenal syndrome in rat. Biomedicine & Pharmacotherapy 2019, 109, 658-670, 10.1016/j.biopha.2018.10.095.
- Milad Ashrafizadeh; Amirhossein Sahebkar; Stephen L. Atkin; Amirhossein Sahebkar; Effects of newly introduced antidiabetic drugs on autophagy. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 2019, 13, 2445-2449, 10.1016/j.dsx.2019.06.028.
- Daniel J. Klionsky; Kotb Abdelmohsen; Akihisa Abe; Joynal Abedin; Hagai Abeliovich; Abraham Acevedo-Arozena; Hiroaki Adachi; Christopher M. Adams; Peter D Adams; Khosrow Adeli; et al.Peter J AdhihettySharon G AdlerGalila AgamRajesh AgarwalManish K AghiMaria AgnelloP. AgostinisPatricia V AguilarJulio Aguirre-GhisoEdoardo M AiroldiSlimane Ait-Si-AliTakahiko AkematsuEmmanuel T AkporiayeMohamed Al-RubeaiGuillermo M. AlbaicetaChris AlbaneseDiego AlbaniMatthew L AlbertJesús AldudoHana AlgülMehrdad AlirezaeiIraide AllozaAlexandru AlmasanMaylin Almonte-BecerilEmad S AlnemriCovadonga AlonsoNihal Altan-BonnetDario C AltieriLydia AlvarezLydia Alvarez-ErvitiSandro AlvesGiuseppina AmadoroAtsuo AmanoConsuelo AmantiniSantiago AmbrosioIvano AmelioAmal O AmerMohamed AmessouAngelika AmonZhenyi AnFrank A AnaniaStig U AndersenUsha AndleyCatherine K AndreadiNathalie Andrieu-AbadieAlberto AnelDavid K AnnShailendra Anoopkumar-DukieManuela AntonioliHiroshi AokiNadezda ApostolovaSaveria AquilaKatia AquilanoKoichi ArakiEli AramaAgustín ArandaJun ArayaAlexandre ArcaroEsperanza AriasHirokazu ArimotoAileen R AriosaJane ArmstrongThierry ArnouldIvica ArsovKatsuhiko AsanumaValerie AskanasÉric AsselinRyuichiro AtarashiSally S AthertonJulie D. AtkinLaura D AttardiPatrick AubergerGeorg AuburgerLaure AurelianRiccardo AutelliLaura AvaglianoMaria Laura AvantaggiatiLimor AvrahamiSuresh AwaleNeelam AzadTiziana BachettiJonathan M BackerDong-Hun BaeJae-Sung BaeOk-Nam BaeSoo Han BaeEric H BaehreckeSeung-Hoon BaekStephen BaghdiguianAgnieszka Bagniewska-ZadwornaHua BaiJie BaiXue-Yuan BaiYannick BaillyKithiganahalli Narayanaswamy BalajiWalter BalduiniAndrea BallabioRena BalzanRajkumar BanerjeeGabor BánhegyiHaijun BaoBenoit BarbeauMaria D BarrachinaEsther BarreiroBonnie BartelAlberto BartoloméDiane C BasshamMaria Teresa BassiRobert C. BastAlakananda BasuMaria Teresa BatistaHenri BatokoMaurizio BattinoKyle BauckmanBradley L BaumgarnerK Ulrich BayerRupert BealeJean-Francois BeaulieuGeorge R. BeckChristoph BeckerJ David BeckhamPierre-Andre BedardPatrick J BednarskiThomas J BegleyChristian BehlChristian BehrendsGeorg Mn BehrensKevin E BehrnsEloy BejaranoAmine BelaidFrancesca BelleudiGiovanni BenardGuy BerchemDaniele BergamaschiMatteo BergamiBen BerkhoutLaura BerliocchiAmelie BernardMonique BernardFrancesca BernassolaAnne BertolottiAmanda S BessSébastien BesteiroSaverio BettuzziSavita BhallaShalmoli BhattacharyyaSujit K BhutiaCaroline BiagoschMichele Wolfe BianchiMartine Biard-PiechaczykViktor BillesCláudia BincolettoBaris BingolSara W BirdMarc BitounIvana BjedovCraig BlackstoneLionel BlancGuillermo A BlancoHeidi Kiil BlomhoffEmilio Boada RomeroStefan BöcklerMarianne BoesKathleen Boesze-BattagliaLawrence H BoiseAlessandra BolinoAndrea BomanPaolo BonaldoMatteo BordiJurgen BoschLuis M BotanaJoëlle BottiGermán BouMarina BouchéMarion BouchecareilhMarie-Josee BoucherMichael E BoultonSebastien G. BouretPatricia BoyaMichaël Boyer-GuittautPeter V. BozhkovNathan BradyVania Mm BragaClaudio BrancoliniGerhard H. BrausJose Manuel Bravo-San PedroLisa A BrennanEmery H BresnickPatrick BrestDave BridgesMarie-Agnès BringerMarisa BriniGlauber C BritoBertha BrodinPaul S BrookesEric J BrownKaren BrownHal E BroxmeyerAlain BruhatPatrícia Chakur BrumJohn H BrumellNicola Brunetti-PierriRobert J. Bryson-RichardsonShilpa BuchAlastair M BuchanHikmet BudakDmitry V BulavinScott J BultmanGeert BultynckVladimir BumbasirevicYan BurelleRobert E BurkeMargit BurmeisterPeter BütikoferLaura CaberlottoKen CadwellMonika CahovaNgsheng CaiJingjing CaiQian CaiSara CalatayudNadine CamougrandMichelangelo CampanellaGrant R. CampbellMatthew CampbellSilvia CampelloRobin CandauIsabella CaniggiaLavinia CantoniLizhi CaoAllan B CaplanMichele CaragliaClaudio CardinaliSandra Morais CardosoJennifer S CarewLaura A CarletonCathleen R CarlinSilvia CarloniSven R. CarlssonDidac Carmona-GutierrezLeticia Am CarneiroOliana CarnevaliSerena CarraAlice CarrierBernadette CarrollCaty CasasJosefina CasasGiuliana CassinelliPerrine CastetsSusana Castro-ObregónGabriella CavalliniI. CeccheriniFrancesco CecconiArthur I CederbaumValentín CeñaSimone CenciClaudia CerellaDavide CerviaSilvia CetrulloHassan ChaachouayHan-Jung ChaeAndrei ChaginChee-Yin ChaiGopal ChakrabartiGeorgios ChamilosEdmond Yw ChanMatthew T.V. ChanDhyan ChandraPallavi ChandraMing-Shi ChangRaymond Chuen-Chung ChangTa Yuan ChangJohn C ChathamSaurabh ChatterjeeSantosh ChauhanYongsheng CheMichael E. CheethamRajkumar CheluvappaChun-Jung ChenGang ChenGuang-Chao ChenGuoqiang ChenHongzhuan ChenJeff W ChenJian-Kang ChenMin ChenMingzhou ChenPeiwen ChenQi ChenQuan ChenShang-Der ChenSi ChenSteve S-L ChenWei-Jung ChenWen Qiang ChenWenli ChenXiangmei ChenYau-Hung ChenYe-Guang ChenYi Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1-222, 10.1080/15548627.2015.1100356.
- Saori Yoshii; Noboru Mizushima; Monitoring and Measuring Autophagy. International Journal of Molecular Sciences 2017, 18, 1865, 10.3390/ijms18091865.
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J.. Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, 2000; pp. Section 5.4. Organelles of the Eukaryotic Cell.
- Xiao-Yu Li; Shan-Shan Wang; Zhe Han; Fei Han; Yun-Peng Chang; Yang Yang; Mei Xue; Bei Sun; Liming Chen; Triptolide Restores Autophagy to Alleviate Diabetic Renal Fibrosis through the miR-141-3p/PTEN/Akt/mTOR Pathway. Molecular Therapy - Nucleic Acids 2017, 9, 48-56, 10.1016/j.omtn.2017.08.011.
- Wei Xin; Zhaoping Li; Ying Xu; Yue Yu; Qi Zhou; Liyong Chen; Qiang Wan; Autophagy protects human podocytes from high glucose-induced injury by preventing insulin resistance. Metabolism 2016, 65, 1307-1315, 10.1016/j.metabol.2016.05.015.
- Atsuko Tagawa; Mako Yasuda; Shinji Kume; Kosuke Yamahara; Jun Nakazawa; Masami Chin-Kanasaki; Hisazumi Araki; Shin-Ichi Araki; Daisuke Koya; Katsuhiko Asanuma; et al.Eun-Hee KimM. HanedaNobuyuki KajiwaraKazuyuki HayashiHiroshi OhashiSatoshi UgiHiroshi MaegawaTakashi Uzu Impaired Podocyte Autophagy Exacerbates Proteinuria in Diabetic Nephropathy. Diabetes 2015, 65, 755-767, 10.2337/db15-0473.
- Yu Liu; Jia Zhang; Yangjia Wang; Xiangjun Zeng; Apelin involved in progression of diabetic nephropathy by inhibiting autophagy in podocytes.. Cell Death & Disease 2017, 8, e3006-e3006, 10.1038/cddis.2017.414.
- Gautam Runwal; Eleanna Stamatakou; Farah H. Siddiqi; Claudia Puri; Ye Zhu; David C. Rubinsztein; LC3-positive structures are prominent in autophagy-deficient cells.. Scientific Reports 2019, 9, 10147, 10.1038/s41598-019-46657-z.
- Chenke Xu; Wei Wang; Jin Zhong; Fan Lei; Naihan Xu; Yaou Zhang; Weidong Xie; Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochemical Pharmacology 2018, 152, 45-59, 10.1016/j.bcp.2018.03.013.
- Jianguang Gong; Huifang Zhan; Yiwen Li; Wei Zhang; Juan Jin; Qiang He; Krüppel‑like factor 4 ameliorates diabetic kidney disease by activating autophagy via the mTOR pathway.. Molecular Medicine Reports 2019, 20, 3240-3248, 10.3892/mmr.2019.10585.
- Eun-Jung Lee; Min-Kyung Kang; Yun-Ho Kim; Dong Yeon Kim; Hyeongjoo Oh; Soo-Il Kim; Su Yeon Oh; Young-Hee Kang; Dietary Chrysin Suppresses Formation of Actin Cytoskeleton and Focal Adhesion in AGE-Exposed Mesangial Cells and Diabetic Kidney: Role of Autophagy. Nutrients 2019, 11, 127, 10.3390/nu11010127.
- Chenglong Dong; Haining Zheng; Shanshan Huang; Na You; Jiarong Xu; Xiaolong Ye; Qun Zhu; Yamin Feng; Qiang You; Heng Miao; Dafa Ding; Yibing Lu; Heme oxygenase-1 enhances autophagy in podocytes as a protective mechanism against high glucose-induced apoptosis. Experimental Cell Research 2015, 337, 146-159, 10.1016/j.yexcr.2015.04.005.
- Honglei Guo; Bin Wang; Hongmei Li; Lilu Ling; Jianying Niu; Yong Gu; Glucagon-like peptide-1 analog prevents obesity-related glomerulopathy by inhibiting excessive autophagy in podocytes. American Journal of Physiology-Renal Physiology 2018, 314, F181-F189, 10.1152/ajprenal.00302.2017.
- X Zhao; G Liu; H Shen; B Gao; X Li; J Fu; J Zhou; Q Ji; Liraglutide inhibits autophagy and apoptosis induced by high glucose through GLP-1R in renal tubular epithelial cells.. International Journal of Molecular Medicine 2014, 35, 684-92, 10.3892/ijmm.2014.2052.
- Yamin Feng; Sheng Chen; Jiarong Xu; Qun Zhu; Xiaolong Ye; Dafa Ding; Weihao Yao; Yibing Lu; Dysregulation of lncRNAs GM5524 and GM15645 involved in high‑glucose‑induced podocyte apoptosis and autophagy in diabetic nephropathy.. Molecular Medicine Reports 2018, 18, 3657-3664, 10.3892/mmr.2018.9412.
- Olivia Lenoir; Magali Jasiek; Carole Henique; Léa Guyonnet; Björn Hartleben; Tillmann Bork; Anna Chipont; Kathleen Flosseau; Imane Bensaada; Alain Schmitt; Jean-Marc Massé; Michèle Souyri; Tobias B. Huber; Pierre-Louis Tharaux; Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015, 11, 1130-1145, 10.1080/15548627.2015.1049799.
- Ji Hee Lim; Hyung Wook Kim; Min Young Kim; Tae Woo Kim; Eun Nim Kim; Yaeni Kim; Sungjin Chung; Young Soo Kim; Bum Soon Choi; Yong-Soo Kim; Yoon Sik Chang; Hye Won Kim; Cheol Whee Park; Cinacalcet-mediated activation of the CaMKKβ-LKB1-AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy.. Cell Death & Disease 2018, 9, 270, 10.1038/s41419-018-0324-4.
- Ken Inoki; mTOR signaling in autophagy regulation in the kidney.. Seminars in Nephrology 2013, 34, 2-8, 10.1016/j.semnephrol.2013.11.002.
- Stéphanie De Rechter; Jean-Paul Decuypere; Ekaterina Ivanova; Lambertus P. Van Den Heuvel; Humbert De Smedt; Elena Levtchenko; Djalila Mekahli; Autophagy in renal diseases. Pediatric Nephrology 2015, 31, 737-752, 10.1007/s00467-015-3134-2.
- Lili Zhou; Youhua Liu; Wnt/β-catenin signalling and podocyte dysfunction in proteinuric kidney disease. Nature Reviews Nephrology 2015, 11, 535-545, 10.1038/nrneph.2015.88.
- Yan Ding; Mary E. Choi; Regulation of autophagy by TGF-β: emerging role in kidney fibrosis.. Seminars in Nephrology 2013, 34, 62-71, 10.1016/j.semnephrol.2013.11.009.
- Haoran Dai; Qingquan Liu; Baoli Liu; Research Progress on Mechanism of Podocyte Depletion in Diabetic Nephropathy. Journal of Diabetes Research 2017, 2017, 1-10, 10.1155/2017/2615286.
- Munehiro Kitada; Yoshio Ogura; Itaru Monno; Daisuke Koya; Regulating Autophagy as a Therapeutic Target for Diabetic Nephropathy. Current Diabetes Reports 2017, 17, , 10.1007/s11892-017-0879-y.
- Milton Packer; Interplay of adenosine monophosphate‐activated protein kinase/sirtuin‐1 activation and sodium influx inhibition mediates the renal benefits of sodium‐glucose co‐transporter‐2 inhibitors in type 2 diabetes: A novel conceptual framework. Diabetes, Obesity and Metabolism 2020, 22, 734-742, 10.1111/dom.13961.
- Xingchen Zhao; Yuanhan Chen; Xiaofan Tan; Li Zhang; Hong Zhang; Zhilian Li; Shuangxin Liu; Ruizhao Li; Ting Lin; Ruyi Liao; Qianmei Zhang; Wei Dong; Wei Shi; Xinling Liang; Advanced glycation end‐products suppress autophagic flux in podocytes by activating mammalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. The Journal of Pathology 2018, 245, 235-248, 10.1002/path.5077.
- Mai Shi; Shuang Yang; Xinwang Zhu; Da Sun; Dan Sun; Xue Jiang; Congxiao Zhang; Lining Wang; The RAGE/STAT5/autophagy axis regulates senescence in mesangial cells.. Cellular Signalling 2019, 62, 109334, 10.1016/j.cellsig.2019.05.019.
- Shoaib Ahmad Malik; Guillermo Mariño; Aména Ben Younes; Shensi Shen; Francis Harper; Maria Chiara Maiuri; Guido Kroemer; Neuroendocrine regulation of autophagy by leptin. Cell Cycle 2011, 10, 2917-2923, 10.4161/cc.10.17.17067.
- Bingxuan Wang; Charukeshi Chandrasekera P.; John J. Pippin; Leptin- and Leptin Receptor-Deficient Rodent Models: Relevance for Human Type 2 Diabetes. Current Diabetes Reviews 2014, 10, 131-145, 10.2174/1573399810666140508121012.
- Irena Audzeyenka; Dorota Rogacka; Agnieszka Piwkowska; Stefan Angielski; Maciej Jankowski; Viability of primary cultured podocytes is associated with extracellular high glucose-dependent autophagy downregulation.. Molecular and Cellular Biochemistry 2017, 430, 11-19, 10.1007/s11010-017-2949-5.
- Coward, R.; Fornoni, A. Insulin signaling: Implications for podocyte biology in diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 104–110, doi:10.1097/MNH.0000000000000078.
- Johanna Guzman; Alexandra N. Jauregui; Sandra Merscher-Gomez; Ny Maiguel; Cristina Muresan; Alla Mitrofanova; Ana Diez-Sampedro; Joel Szust; Tae-Hyun Yoo; Rodrigo Villarreal; Christopher Pedigo; Ruth Molano; Kevin Johnson; Barbara Kahn; Bjoern Hartleben; Tobias B. Huber; Jharna Saha; George W. Burke; E. Dale Abel; Frank C. Brosius; Alessia Fornoni; Podocyte-Specific GLUT4-Deficient Mice Have Fewer and Larger Podocytes and Are Protected From Diabetic Nephropathy. Diabetes 2014, 63, 701-714, 10.2337/db13-0752.
- Silvia Ezquerro; Gema Frühbeck; Amaia Rodríguez; Ghrelin and autophagy. Current Opinion in Clinical Nutrition and Metabolic Care 2017, 20, 402-408, 10.1097/mco.0000000000000390.
- Zhong-Hui Wang; Wei-Ying Ren; Lei Zhu; Li-Juan Hu; Plasminogen Activator Inhibitor-1 Regulates LPS Induced Inflammation in Rat Macrophages through Autophagy Activation. The Scientific World Journal 2014, 2014, 1-12, 10.1155/2014/189168.
- Yung-Ho Hsu; Hsiao-Chi Chuang; Yu-Hsuan Lee; Yuh-Feng Lin; Yu-Jhe Chiu; Yung-Li Wang; Mai-Szu Wu; Hui-Wen Chiu; Induction of Fibrosis and Autophagy in Kidney Cells by Vinyl Chloride.. Cells 2019, 8, 601, 10.3390/cells8060601.
- Keizo Kanasaki; Emi Kawakita; Daisuke Koya; Relevance of Autophagy Induction by Gastrointestinal Hormones: Focus on the Incretin-Based Drug Target and Glucagon.. Frontiers in Pharmacology 2019, 10, 476, 10.3389/fphar.2019.00476.
- Gur Kaushal; Kiran Chandrashekar; Luis A. Juncos; Sudhir V. Shah; Autophagy Function and Regulation in Kidney Disease. Biomolecules 2020, 10, 100, 10.3390/biom10010100.
- Yushan Zhu; Lixia Zhao; Lei Liu; Ping Gao; Weili Tian; Xiaohui Wang; Haijing Jin; Haidong Xu; Quan Chen; Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein & Cell 2010, 1, 468-477, 10.1007/s13238-010-0048-4.
- Masashi Mizuno; Atsushi Kuno; Toshiyuki Yano; Takayuki Miki; Hiroto Oshima; Tatsuya Sato; Kei Nakata; Yukishige Kimura; Masaya Tanno; Tetsuji Miura; Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiological Reports 2018, 6, e13741, 10.14814/phy2.13741.
- Yi Zhou; Huanyuan Wang; Fuli Man; Zhiying Guo; Jiahui Xu; Wenjing Yan; Jiaying Li; Qi Pan; Wen Wang; Sitagliptin Protects Cardiac Function by Reducing Nitroxidative Stress and Promoting Autophagy in Zucker Diabetic Fatty (ZDF) Rats. Cardiovascular Drugs and Therapy 2018, 32, 541-552, 10.1007/s10557-018-6831-9.
- Hiromichi Murase; Atsushi Kuno; Takayuki Miki; Masaya Tanno; Toshiyuki Yano; Hidemichi Kouzu; Satoko Ishikawa; Toshiyuki Tobisawa; Makoto Ogasawara; Keitaro Nishizawa; Tetsuji Miura; Inhibition of DPP-4 reduces acute mortality after myocardial infarction with restoration of autophagic response in type 2 diabetic rats.. Cardiovascular Diabetology 2015, 14, 103, 10.1186/s12933-015-0264-6.
- Gu, Y.; Ma, C.T.; Gu, H.L.; Shi, L.; Tian, X.T.; Xu, W.Q. Sitagliptin improves cardiac function after myocardial infarction through activation of autophagy in streptozotocin-induced diabetic mice. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8973–8983, doi:10.26355/eurrev_201812_16668.
- Wen-Bin Zheng; Jing Zhou; Shasha Song; Wen Kong; Wenfang Xia; Lulu Chen; Tianshu Zeng; Dipeptidyl-Peptidase 4 Inhibitor Sitagliptin Ameliorates Hepatic Insulin Resistance by Modulating Inflammation and Autophagy in ob/ob Mice. International Journal of Endocrinology 2018, 2018, 1-11, 10.1155/2018/8309723.
- Yuliya Sharkovska; Christoph Reichetzeder; Markus Alter; Oleg Tsuprykov; Sebastian Bachmann; Thomas Secher; Thomas Klein; Berthold Hocher; Blood pressure and glucose independent renoprotective effects of dipeptidyl peptidase-4 inhibition in a mouse model of type-2 diabetic nephropathy. Journal of Hypertension 2014, 32, 2211-2223, 10.1097/hjh.0000000000000328.
- Yu. S. Gavrilova; N. P. Bgatova; Vadim V. Klimontov; I. Yu. Ischenko; S. V. Michurina; N. E. Myakina; E. L. Zavyalov; Effect of Linagliptin on Structural Changes in the Kidney in Experimental Type 2 Diabetes Mellitus. Bulletin of Experimental Biology and Medicine 2016, 161, 501-504, 10.1007/s10517-016-3447-6.



