 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sun Choi | + 4049 word(s) | 4049 | 2021-06-08 12:17:43 |
Video Upload Options
The new advances in deep learning methods have influenced many aspects of scientific research, including the study of the protein system. The prediction of proteins’ 3D structural components is now heavily dependent on machine learning techniques that interpret how protein sequences and their homology govern the inter-residue contacts and structural organization. Especially, methods employing deep neural networks have had a significant impact on recent CASP13 and CASP14 competition. Here, we explore the recent applications of deep learning methods in the protein structure prediction area. We also look at the potential opportunities for deep learning methods to identify unknown protein structures and functions to be discovered and help guide drug–target interactions. Although significant problems still need to be addressed, we expect these techniques in the near future to play crucial roles in protein structural bioinformatics as well as in drug discovery.
1. Introduction
2. Protein Sequence Homology, 3D Structure, and Deep Learning
2.1. Protein Sequence Homology
2.2. 3D Structural Space of Proteins
2.3. Overview of Deep Learning Methods
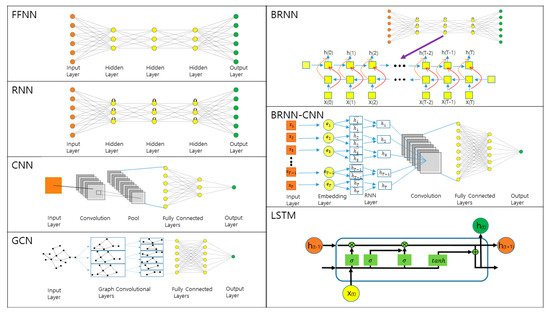
3. Prediction of 1D and 2D Protein Structural Annotations
3.1. 1D Prediction
3.2. 2D Prediction
References
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.-W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D 2019, 75, 861–877.
- Bai, X.-C.; McMullan, G.; Scheres, S.H. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57.
- Wüthrich, K. The way to NMR structures of proteins. Nat. Struct. Biol. 2001, 8, 923–925.
- Drenth, J. Principles of Protein X-ray Crystallography; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2007.
- Anfinsen, C.B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223–230.
- Pauling, L.; Corey, R.B. Configurations of Polypeptide Chains With Favored Orientations Around Single Bonds. Two New Pleated Sheets 1951, 37, 729–740.
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211.
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333.
- Cheng, J.; Tegge, A.N.; Baldi, P. Machine Learning Methods for Protein Structure Prediction. IEEE Rev. Biomed. Eng. 2008, 1, 41–49.
- Sun, S. Reduced representation model of protein structure prediction: Statistical potential and genetic algorithms. Protein Sci. 1993, 2, 762–785.
- Torrisi, M.; Pollastri, G.; Le, Q. Deep learning methods in protein structure prediction. Comput. Struct. Biotechnol. J. 2020, 18, 1301–1310.
- Rost, B.; Sander, C. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins Struct. Funct. Bioinform. 1994, 19, 55–72.
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697.
- Owens, J.D.; Houston, M.; Luebke, D.; Green, S.; Stone, J.E.; Phillips, J.C. GPU computing. Proc. IEEE 2008, 96, 879–899.
- Wilkins, A.D.; Bachman, B.J.; Erdin, S.; Lichtarge, O. The use of evolutionary patterns in protein annotation. Curr. Opin. Struct. Biol. 2012, 22, 316–325.
- Floudas, C.; Fung, H.; McAllister, S.; Mönnigmann, M.; Rajgaria, R. Advances in protein structure prediction and de novo protein design: A review. Chem. Eng. Sci. 2006, 61, 966–988.
- Moult, J. A decade of CASP: Progress, bottlenecks and prognosis in protein structure prediction. Curr. Opin. Struct. Biol. 2005, 15, 285–289.
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)—Round XII. Proteins Struct. Funct. Bioinform. 2018, 86, 7–15.
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)-Round XIII. Proteins 2019, 87, 1011–1020.
- Sun, T.; Zhou, B.; Lai, L.; Pei, J. Sequence-based prediction of protein protein interaction using a deep-learning algorithm. BMC Bioinform. 2017, 18, 1–8.
- Wen, M.; Zhang, Z.; Niu, S.; Sha, H.; Yang, R.; Yun, Y.; Lu, H. Deep-learning-based drug–target interaction prediction. J. Proteome Res. 2017, 16, 1401–1409.
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637.
- Rodionov, M.A.; Blundell, T.L. Sequence and structure conservation in a protein core. Proteins Struct. Funct. Bioinform. 1998, 33, 358–366.
- Sadowski, M.I.; Jones, D.T. The sequence–structure relationship and protein function prediction. Curr. Opin. Struct. Biol. 2009, 19, 357–362.
- Schmidhuber, J. Deep learning in neural networks: An overview. Neural Netw. 2015, 61, 85–117.
- Werbos, P.J. Backpropagation through time: What it does and how to do it. Proc. IEEE 1990, 78, 1550–1560.
- Hochreiter, S.; Bengio, Y.; Frasconi, P.; Schmidhuber, J. Gradient flow in recurrent nets: The difficulty of learning long-term dependencies. In A Field Guide to Dynamical Recurrent Neural Networks; IEEE Press: Hoboken, NJ, USA, 2001.
- Minai, A.A.; Williams, R.D. Perturbation response in feedforward networks. Neural Netw. 1994, 7, 783–796.
- Szegedy, C.; Ioffe, S.; Vanhoucke, V.; Alemi, A. Inception-v4, Inception-Resnet and the Impact of Residual Connections on Learning. In Proceedings of the AAAI Conference on Artificial Intelligence, San Francisco, CA, USA, 4–9 February 2017.
- Hu, Y.; Huber, A.; Anumula, J.; Liu, S.-C. Overcoming the vanishing gradient problem in plain recurrent networks. arXiv 2018, arXiv:1801.06105.
- Schuster, M.; Paliwal, K.K. Bidirectional recurrent neural networks. IEEE Trans. Signal. Process. 1997, 45, 2673–2681.
- Baldi, P.; Brunak, S.; Frasconi, P.; Soda, G.; Pollastri, G. Exploiting the past and the future in protein secondary structure prediction. Bioinformatics 1999, 15, 937–946.
- Di Lena, P.; Nagata, K.; Baldi, P. Deep architectures for protein contact map prediction. Bioinformatics 2012, 28, 2449–2457.
- Pérez-Ortiz, J.A.; Gers, F.A.; Eck, D.; Schmidhuber, J. Kalman filters improve LSTM network performance in problems unsolvable by traditional recurrent nets. Neural Netw. 2003, 16, 241–250.
- LeCun, Y.; Boser, B.; Denker, J.S.; Henderson, D.; Howard, R.E.; Hubbard, W.; Jackel, L.D. Backpropagation Applied to Handwritten Zip Code Recognition. Neural Comput. 1989, 1, 541–551.
- Yin, W.; Kann, K.; Yu, M.; Schütze, H. Comparative study of CNN and RNN for natural language processing. arXiv 2017, arXiv:1702.01923.
- Hanson, J.; Paliwal, K.; Litfin, T.; Yang, Y.; Zhou, Y. Accurate prediction of protein contact maps by coupling residual two-dimensional bidirectional long short-term memory with convolutional neural networks. Bioinformatics 2018, 34, 4039–4045.
- Gligorijevic, V.; Renfrew, P.D.; Kosciolek, T.; Leman, J.K.; Berenberg, D.; Vatanen, T.; Chandler, C.; Taylor, B.C.; Fisk, I.M.; Vlamakis, H. Structure-based function prediction using graph convolutional networks. bioRxiv 2020.
- Torrisi, M.; Kaleel, M.; Pollastri, G. Deeper profiles and cascaded recurrent and convolutional neural networks for state-of-the-art protein secondary structure prediction. Sci. Rep. 2019, 9, 1–12.
- Zhang, Y.; Qiao, S.; Ji, S.; Li, Y. DeepSite: Bidirectional LSTM and CNN models for predicting DNA–protein binding. Int. J. Mach. Learn. Cybern. 2020, 11, 841–851.
- Yang, Y.; Gao, J.; Wang, J.; Heffernan, R.; Hanson, J.; Paliwal, K.; Zhou, Y. Sixty-five years of the long march in protein secondary structure prediction: The final stretch? Brief. Bioinform. 2018, 19, 482–494.
- Cuff, J.A.; Clamp, M.E.; Siddiqui, A.S.; Finlay, M.; Barton, G.J. JPred: A consensus secondary structure prediction server. Bioinformatics 1998, 14, 892–893.
- Cuff, J.A.; Barton, G.J. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins Struct. Funct. Bioinform. 2000, 40, 502–511.
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405.
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402.
- Magnan, C.N.; Baldi, P. SSpro/ACCpro 5: Almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 2014, 30, 2592–2597.
- Bau, D.; Martin, A.J.; Mooney, C.; Vullo, A.; Walsh, I.; Pollastri, G. Distill: A suite of web servers for the prediction of one-, two- and three-dimensional structural features of proteins. BMC Bioinform. 2006, 7, 402.
- Torrisi, M.; Kaleel, M.; Pollastri, G. Porter 5: Fast, state-of-the-art ab initio prediction of protein secondary structure in 3 and 8 classes. bioRxiv 2018.
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175.
- Mooney, C.; Vullo, A.; Pollastri, G. Protein structural motif prediction in multidimensional ø-ψ space leads to improved secondary structure prediction. J. Comput. Biol. 2006, 13, 1489–1502.
- Kaleel, M.; Torrisi, M.; Mooney, C.; Pollastri, G. PaleAle 5.0: Prediction of protein relative solvent accessibility by deep learning. Amino Acids 2019, 51, 1289–1296.
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sønderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins Struct. Funct. Bioinform. 2019, 87, 520–527.
- Wood, M.J.; Hirst, J.D. Protein secondary structure prediction with dihedral angles. PROTEINS Struct. Funct. Bioinform. 2005, 59, 476–481.
- Kountouris, P.; Hirst, J.D. Prediction of backbone dihedral angles and protein secondary structure using support vector machines. BMC Bioinform. 2009, 10, 1–14.
- Faraggi, E.; Zhang, T.; Yang, Y.; Kurgan, L.; Zhou, Y. SPINE X: Improving protein secondary structure prediction by multistep learning coupled with prediction of solvent accessible surface area and backbone torsion angles. J. Comput. Chem. 2012, 33, 259–267.
- Hanson, J.; Paliwal, K.; Litfin, T.; Yang, Y.; Zhou, Y. Improving prediction of protein secondary structure, backbone angles, solvent accessibility and contact numbers by using predicted contact maps and an ensemble of recurrent and residual convolutional neural networks. Bioinformatics 2019, 35, 2403–2410.
- Yang, Y.; Heffernan, R.; Paliwal, K.; Lyons, J.; Dehzangi, A.; Sharma, A.; Wang, J.; Sattar, A.; Zhou, Y. Spider2: A package to predict secondary structure, accessible surface area, and main-chain torsional angles by deep neural networks. In Prediction of Protein Secondary Structure; Springer: Berlin/Heidelberg, Germany, 2017; pp. 55–63.
- Heffernan, R.; Yang, Y.; Paliwal, K.; Zhou, Y. Capturing non-local interactions by long short-term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure, backbone angles, contact numbers and solvent accessibility. Bioinformatics 2017, 33, 2842–2849.
- Kotowski, K.; Smolarczyk, T.; Roterman-Konieczna, I.; Stapor, K. ProteinUnet—An efficient alternative to SPIDER3-single for sequence-based prediction of protein secondary structures. J. Comput. Chem. 2021, 42, 50–59.
- Heffernan, R.; Paliwal, K.; Lyons, J.; Singh, J.; Yang, Y.; Zhou, Y. Single-sequence-based prediction of protein secondary structures and solvent accessibility by deep whole-sequence learning. J. Comput. Chem. 2018, 39, 2210–2216.
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59.
- Dosztányi, Z. Prediction of protein disorder based on IUPred. Protein Sci. 2018, 27, 331–340.
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863.
- Hanson, J.; Paliwal, K.K.; Litfin, T.; Zhou, Y. SPOT-Disorder2: Improved Protein Intrinsic Disorder Prediction by Ensembled Deep Learning. Genom. Proteom. Bioinform. 2019, 17, 645–656.
- Hanson, J.; Yang, Y.; Paliwal, K.; Zhou, Y. Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics 2017, 33, 685–692.
- Aszodi, A.; Gradwell, M.; Taylor, W. Global fold determination from a small number of distance restraints. J. Mol. Biol. 1995, 251, 308–326.
- Kim, D.E.; DiMaio, F.; Yu-Ruei Wang, R.; Song, Y.; Baker, D. One contact for every twelve residues allows robust and accurate topology-level protein structure modeling. Proteins Struct. Funct. Bioinform. 2014, 82, 208–218.
- Bitbol, A.-F. Inferring interaction partners from protein sequences using mutual information. PLoS Comput. Biol. 2018, 14, e1006401.
- Morcos, F.; Pagnani, A.; Lunt, B.; Bertolino, A.; Marks, D.S.; Sander, C.; Zecchina, R.; Onuchic, J.N.; Hwa, T.; Weigt, M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA 2011, 108, E1293–E1301.
- Jones, D.T.; Buchan, D.W.; Cozzetto, D.; Pontil, M. PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics 2012, 28, 184–190.
- Edgar, R.C.; Batzoglou, S. Multiple sequence alignment. Curr. Opin. Struct. Biol. 2006, 16, 368–373.
- Dos Santos, R.N.; Morcos, F.; Jana, B.; Andricopulo, A.D.; Onuchic, J.N. Dimeric interactions and complex formation using direct coevolutionary couplings. Sci. Rep. 2015, 5, 1–10.
- Walsh, I.; Bau, D.; Martin, A.J.; Mooney, C.; Vullo, A.; Pollastri, G. Ab initio and template-based prediction of multi-class distance maps by two-dimensional recursive neural networks. BMC Struct Biol. 2009, 9, 5.
- Eickholt, J.; Cheng, J.L. A study and benchmark of DNcon: A method for protein residue-residue contact prediction using deep networks. BMC Bioinform. 2013, 14, 1–10.
- Jones, D.T.; Singh, T.; Kosciolek, T.; Tetchner, S. MetaPSICOV: Combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics 2015, 31, 999–1006.
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLoS Comput. Biol. 2017, 13, e1005324.
- Adhikari, B.; Hou, J.; Cheng, J.L. DNCON2: Improved protein contact prediction using two-level deep convolutional neural networks. Bioinformatics 2018, 34, 1466–1472.
- Li, Y.; Zhang, C.X.; Bell, E.W.; Zheng, W.; Zhou, X.G.; Yu, D.J.; Zhang, Y.; Kolodny, R. Deducing high-accuracy protein contact-maps from a triplet of coevolutionary matrices through deep residual convolutional networks. PLoS Comput. Biol. 2021, 17, e1008865.
- Liu, Y.; Palmedo, P.; Ye, Q.; Berger, B.; Peng, J. Enhancing Evolutionary Couplings with Deep Convolutional Neural Networks. Cell Syst. 2018, 6, 65–74.e3.
- Jones, D.T.; Kandathil, S.M. High precision in protein contact prediction using fully convolutional neural networks and minimal sequence features. Bioinformatics 2018, 34, 3308–3315.
- Michel, M.; Menendez Hurtado, D.; Elofsson, A. PconsC4: Fast, accurate and hassle-free contact predictions. Bioinformatics 2019, 35, 2677–2679.
- Ji, S.; Oruc, T.; Mead, L.; Rehman, M.F.; Thomas, C.M.; Butterworth, S.; Winn, P.J. DeepCDpred: Inter-residue distance and contact prediction for improved prediction of protein structure. PLoS ONE 2019, 14, e0205214.
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710.


