 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anna Gliszczyńska | + 3246 word(s) | 3246 | 2021-04-26 04:13:47 | | | |
| 2 | Peter Tang | Meta information modification | 3246 | 2021-06-23 11:53:52 | | |
Video Upload Options
S-(+) enantiomer of ibuprofen (IBU) dexibuprofen (DXI) is known to be more potent than its R-(−) form and exhibits many advantages over the racemic mixture of IBU such as lower toxicity, greater clinical efficacy, and lesser variability in therapeutic effects. Moreover, DXI potential has been recently advocated to reduce cancer development and prevent the development of neurodegenerative diseases in addition to its anti-inflammatory properties.
1. Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are members of a drug class that reduces pain, decreases fever, prevents blood clots, and decreases inflammation [1][2]. In general, NSAIDs are characterized by a high degree of protein binding and small volumes of distribution [3]. However, in this family, differences in clearance and variability in half-life are of special relevance. Among of all NSAIDs, ibuprofen (IBU) is one of the most widely and frequently employed therapeutic agents with the lowest toxicity. IBU was initially introduced in the UK in 1960s and afterwards during the 1970s worldwide as a prescription-only medication with recommendation dose 2400 mg/day (or higher in the USA) [4]. Nowadays, IBU is widely used in many countries and presents minimum side effects involving gastric damage observed in many other NSAIDs. Over the last three decades, the over-the-counter (OTC) IBU-containing preparations and the generic preparations sold by prescription and non-prescription are used as a NSAID with analgesic and antipyretic properties [5]. They are used for the treatment of osteoarthritis, rheumatoid arthritis and in a wide variety of other painful conditions [6]. These therapeutic effects come from activity of IBU to block prostaglandin synthesis by a non-selective, reversible inhibition of the cyclooxygenase enzymes COX-1 and COX-2 [7]. IBU achieves high plasma protein binding and low distribution volume, but it possesses the ability to be accumulated in effective amounts in compartments where inflammation occurs. These compartments, such as cerebrospinal fluid (CSF) where there is need for anti-inflammatory/analgesic activity, are the target of IBU.
In most of the commercially available preparations, IBU occurs only as a diastereoisomeric mixture which comprises equal quantities of R-(−)-ibuprofen and S-(+)-ibuprofen. Dextrarotatory isomer of ibuprofen dexibuprofen (DXI) is the pharmacologically effective enantiomer, which was for the first time launched in Austria in 1994 [8]. Racemic ibuprofen and DXI differ in their physical, chemical, and pharmacological properties as well as their metabolic profile [5][9]. In the last five years, 4836 patients have been exposed to DXI in clinical trials and post marketing surveillance (PMS) trials. Only in 3.7% of patients have adverse drug reactions been reported and three serious adverse drug reactions (0.06%) occurred [5]. It has been proven in the in vitro model that dextrarotatory isomer exhibits about 160-times higher activity in prostaglandins inhibition in comparison to enantiomer (R). Other studies of thromboxane generation in clotting blood also confirmed higher activity of enantiomer S than racemate [10]. Therefore, it would be highly advantageous to use DXI as a pain reliver. Especially, due to the fact that while DXI effectively inhibits the activity of COX-1 and COX-2, the enantiomer (R) demonstrates the inhibition only towards COX-1 and it is worth noting that it is responsible increasing the side effects in the gastrointestinal tract [11].
rac-Ibuprofen undergoes an unusual metabolic fate because inactive R-(−)-ibuprofen is enzymatically converted to the therapeutically active enantiomer S-(+) [12]. This may contribute to variability in analgesia including delayed onset of activity and may explain the poor relationship observed between plasma concentrations of IBU and clinical response for acute pain and rheumatoid arthritis [13]. According to published data in humans, average 50% of enantiomer (−) is metabolically converted via catalytic activity of fatty acyl coenzyme thioesterase to more active enantiomer (+) [14][15] in the intestinal tract and liver after oral absorption [16][17]. However, it should be pointed out that the level of metabolic inversion of IBU may vary between 35 and 85% depending on the formulation type, condition of the liver, the intake of medicines, and the state of the disease [18][19]. During this process, the first step is the activation of R-(−)-ibuprofen in the presence of coenzyme A (CoA), adenosine triphosphate (ATP), and Mg2+ (Figure 1).
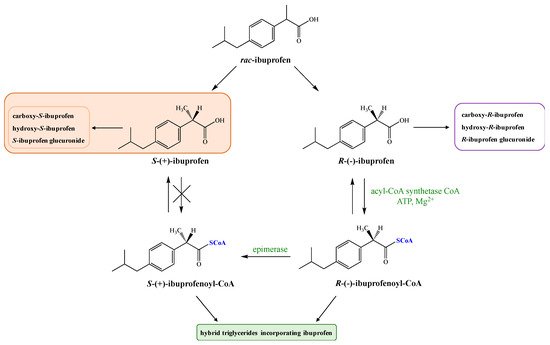
Next, the AMP-derivative is esterified with coenzyme A (CoA) by the action of acyl-CoA synthetase. R-(−)-ibuprofen-CoA undergoes epimerization via the actions of the -methylacyl-coenzyme A racemase (encoded by gene AMACR) [20] to form S-(+)-ibuprofen-CoA, which is then hydrolyzed by a hydrolase to form S-(+)-ibuprofen [5]. The key step of this process is the removal of the substrate alfa-proton followed by non-stereoselective reprotonation. The process of inversion can occur pre-systematically in the gut [21] as well as in the liver [22].
Thioesters formed from coenzyme A and ibuprofen are subsequently incorporated into triglycerides (TAG) or phospholipids (PLs), forming hybrids that may have an influence on the function of the cell membranes. However, it is described in the literature that only R enantiomer of IBU has been detected in the human adipose tissue and its elimination half-life (t1/2) is about seven days. It was proven that oral administration of pure enantiomer S allows a stronger analgesic effect in a shorter time compared with racemic mixture. Higher effectiveness therapy of S-(+)-ibuprofen and significantly reduced side effects of its administration have been demonstrated on the group of 1400 patients [21][23].
As a general approach, it was firstly claimed that a dose of 1:0.75 (ibuprofen and DXI, respectively) would be necessary in order to obtain similar pharmaceutical efficacy with DXI than IBU [5]. Regarding this aspect, several clinical trials have been performed using a dose ratio of 0.5:1 assuming the fact that 50% of the dose contained in IBU corresponds to DXI. In these clinical trials, DXI therapeutic efficacy was similar to IBU. Other studies carried out comparing DXI with other NSAID such as Diclofenac pointed out that regarding the maximum daily dose (MDD) using 75% of the MDD of DXI, the same efficacy as 100% of Diclofenac was achieved [8]. Moreover, regarding DXI interactions with other compounds, the data obtained to date indicate that no influence was found on bioavailability with or without food intake. So far, DXI has demonstrated clinical efficacy in rheumatoid arthritis, ankylosing spondylitis, osteoarthritis of the hip, osteoarthritis of the knee, lumbar vertebral syndrome, distortion of the ankle joint and dysmenorrhoea. The tolerability was better than other NSAIDS since racemic ibuprofen showed a 30% increasing incidence of adverse drug reactions and diclofenac a 90% higher than DXI. Therefore, DXI would potentially combine the high efficacy of diclofenac with the good tolerability of ibuprofen [8]. In addition, DXI has also been compared with Celecoxib, a selective inhibitor of cyclooxygenase-2 (COX-2) for the treatment of hip osteoarthritis. DXI demonstrated equal efficacy to Celecoxib and a comparable tolerability profile [24].
DXI has a slower dissolution rate in the simulated gastric and enteric juices compared with the racemic ibuprofen and displays improved oral bioavailability. Pure DXI exhibits more advantages than its former, but due to its weak acidity (pKa = 5.2) unfortunately its bioavailability is relatively low due to the limited solubility in acidic media of the stomach. According to Biopharmaceutics Classification System (BCS), DXI is classified as the poorly water-soluble drug but well-penetrating substances substance (group II). Therefore, its bioavailability is limited by its poor dissolution, so its absorption rate is similar to that of dissolution. These physicochemical properties of DXI result in many limitations in in vivo models like incomplete release, food interactions, or high inter-subject variability. To overcome these mentioned limitations, many attempts have been made so far.
2. Dexibuprofen Main Pharmacological Properties
Dexibuprofen, S-(+)-ibuprofen, (2S)-2-(4-isobutylphenyl)propionic acid (DXI), is the dextrarotatory, more pharmacologically effective and more potent enantiomer of ibuprofen [25]. Enantiomer S-(+) IBU exerts its effect by suppressing the prostanoid synthesis in the inflammatory cells mainly via inhibition of the COX-2 isoform, causing analgesic, antipyretic, and anti-inflammatory effects [10][26]. S-(+)-ibuprofen is less toxic than the enantiomer R. DXI possesses higher water solubility (68.4 mg/L) than ibuprofen (21 mg/L) and a lower melting point (49–53 °C vs 75–77.5 °C, DXI and ibuprofen, respectively). Moreover, DXI also presents increased stability, a slower dissolution rate, and improved bioavailability [26].
DXI is currently available in the European Union (EU) currently manufactured as tablets against pain and inflammation containing 400 mg of DXI [27]. Moreover, several clinical trials are underway. In the US data base, two Phase I clinical trials with a currently unknown status have been approved to evaluate the safety of DXI 300 mg and 200 mg after a single and multiple dose oral administration in healthy participants (NCT0295651, NCT02956525) [28][29]. Moreover, a completed phase I clinical trial has also been performed by Gebro Pharma GmbH to assess DXI effects on aspirin-treated volunteers (NCT00442585) [29]. Furthermore, in 2010 a phase 3 clinical trial was completed aimed at studying the efficacy and safety of DXI syrup with fever due to common cold (NCT00812422) [30]. A phase IV postmarked clinical study completed in 2012, assessed and compared the tolerability profile of DXI Gebro 400 mg powder for oral suspension against ibuprofen 400 mg in patients with painful osteoarthritis of the hip or knee (NCT01066676) [31]. In the EU database, a completed prospective phase 3 clinical trial is registered, aimed at investigating the safety, tolerability and efficacy of DXI Gebro 400 mg powder for oral suspension (test) compared to ibuprofen 400 mg powder for oral suspension (reference) in patients suffering from osteoarthritis of the hip or knee, showing positive results for DXI against its racemic counterpart (2009-010719-33) [32].
The majority of DXI formulations are intended to be administered orally to reach systemic circulation [33]. After oral administration, DXI is rapidly and extensively absorbed from the upper GI tract with its peak values in the plasma and serum at approximately tmax, ~ 1–2 h depending on the specific oral formulations [5]. Moreover, it has been reported that unbound DXI concentrations show linear pharmacokinetics at commonly used doses. Absorption of DXI occurs on a principle of a passive process where its crystal form may play a crucial role for the interpretation of experimental and clinical data. It should be noted that administration of DXI with meals delays the time to reach maximum concentration (from 2.1 h after fasting conditions to 2.8 h after non-fasting conditions) and decreases marginally the maximum plasma concentration (from 20.6 to 18.1 g/mL). However, food has no effect on the extent of absorption. A linear dose-response relationship was shown over the dose range from 200 to 400 mg [14][21][34].
Phase I metabolism of both enantiomers of IBU involves hydroxylation of the isobutyl chains to 2-hydroxy- and 3-hydroxyibuprofen and subsequent oxidation of the latter to 3-carboxyibuprofen and p-carboxy-2-propionate (Figure 2). Moreover, some reports confirm DXI preference against the R-enantiomer in both phase I and II metabolism [35].
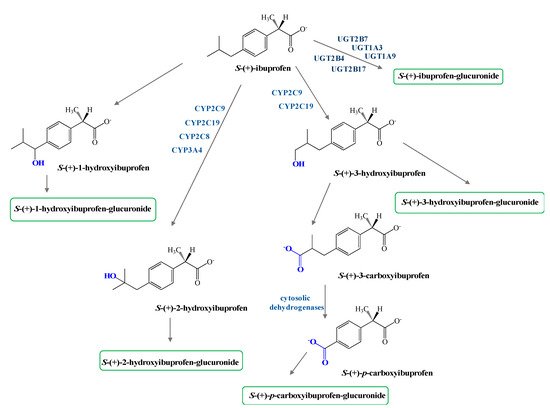
In addition, protein binding has been determined to be enantioselective. In the case of IBU, the R-enantiomer has shown that the binding constant between proteins and the R-enantiomer high-affinity protein binding site is 2.6-fold higher than for the S-enantiomer (DXI). This decreased binding of DXI may cause higher transfer into blister fluid or synovial fluid, where it elicits the pharmacological activity [35][36].
The major metabolites are 2-hydroxy-ibuprofen and carboxy-ibuprofen (and their corresponding acyl glucuronides) which account in the urinary excretion for around 25% and 37% of an administrated dose, respectively [14][21][34], Metabolism of S-(+)-ibuprofen is predominantly catalyzed via cytochrome isoform CYP-2C9, whereas its R-(−) antipode is formed more via isoform CYP-2C8, [36][37]. No difference in the pharmacokinetics of R-(−) and S-(+) enantiomer of IBU have been found related with gender with the exception of the volume of distribution VD/F being about twice that in adult females compared with males [38].
3. Dexibuprofen Prodrugs
NSAIDs are one of the therapeutic groups most widely used, prescribed, and over the counter medications, which administration leads to serious gastrointestinal (GI) complications. These side effects of NSAIDs associated with long term of their oral use, are attributed to the presence of free carboxylic group (-COOH) in their structures. Therefore, many attempts have been made, especially in the last decade, to mask this free carboxylic group and synthesized prodrugs of NSAIDs [39][40] and also overcome pharmaceutical, pharmacokinetic, and pharmacodynamic barriers. In the case of DXI, a number of functional groups have been employed for the preparation of its prodrugs that are trying to reduce its tendency to create peptic ulceration and GI track bleeding. Its conjugates with polymers, amino acids, and ethanolamine have been obtained and proposed as the forms that can constitute an effective approach to solve the problems, slow down DXI action achieving a prolonged drug effect, avoiding adverse effects and increasing therapeutic adherence.
Ashraf et al. synthesized two series of prodrugs of DXI, amides and esters (Figure 3). To synthesize amide prodrugs 1a–e five amino acids were selected: glycine, alanine, valine, leucine, and phenyl alanine. The ester prodrugs 2a–e were obtained in the reaction of DXI with different alcohols: isopropyl, n-butyl, isobutyl, benzyl, and glycerol. The solubility studies showed that synthesized series of DXI derivatives have more lipophilic character and exhibit low protein binding that lead to an increase in their availability in plasma for hydrolysis and reduce the required active dose. All studied prodrugs showed hydrolysis rate in simulated intestinal fluid (SIF pH 7.4) in 80% plasma. It was also proved that DXI in the form of prodrugs prevents the accumulation of the drug in gastric mucosa, decreasing the possible gastrointestinal irritant without loss of pharmacological effect [41].
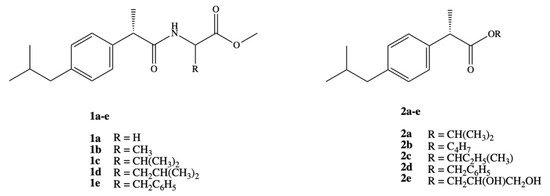
These findings were deeply extended and DXI amide analogues with alkyl/aryl substitution were evaluated as anticancer agents towards breast carcinoma cell line (MCF-7). It was determined that the presence of chlorine atom in the phenyl ring has the strongest effect on biological activity and the amides bearing 2,5-dichloro and 2-chloro substituted phenyl ring turned out to be the most active and exhibited 100% inhibition of the tumor growth. Dichloride substituted amide N-(2,5-dichlorophenyl)-2-(4-isobutylphenyl)-propionamide was active even at lower concentration doses IC50 = 0.01 ± 0.002 μM than doxorubicin IC50 = 0.04 ± 0.006 μM [42]. Amide derivatives of DXI 1b,d,e were also studied in the aspect of how they interact with DNA using spectroscopic, electrochemical, and molecular docking techniques under simulating physiological conditions at stomach and blood pH, 4.7 and 7.4, respectively at temperature 37 °C. The authors observed spontaneous interaction of all the compounds with DNA via intercalation and external bindings [43].
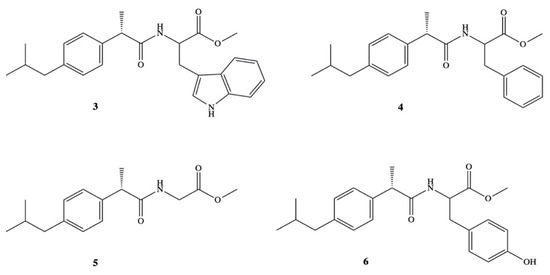
High solubility in organic solvents and high hydrolysis rate in 80% human plasma have been reported also for amide prodrugs of DXI 3–6 synthesized in the reaction of DXI acid chloride with methyl esters of L-tryptophan, L-phenylalanine, glycine and l-tyrosine (Figure 4) [44]. Prodrugs 3–6 were studied on their various physicochemical as well as pharmacological properties on the group of Wistar albino rats. Amides were found to be significantly less ulcerogenic with much higher anti-inflammatory activity (64.5–77.3%) than that reported for the parent drug (43.3%). The maximum of their activity was observed at 6 h and was practically constant up to 8 h.
Rasheed and colleagues synthesized DXI-dextran prodrug 7 for oral administration aimed at improving DXI aqueous solubility, increasing its therapeutic efficacy, and reducing gastrointestinal side effects (Figure 5) [45]. They used DXI acyl imidazole derivatives and dextran of different molecular weights (10,000–20,000 Da) to obtain DXI-dextran prodrugs with the substitution degree between 15.6–16.5%. Obtained prodrugs were characterized by faster hydrolysis at pH 9.0 than pH 7.4. In the in vivo study carried out on albino rats, DXI-dextran did not show improvement of the analgesic activity against DXI but showed a slight increase in the anti-inflammatory potential. The results also revealed that ulcerogenicity of DXI-dextran prodrugs in stomach was reduced compared with free form of this drug and clearly indicate that side effects of DXI were reduced after conjugation with dextran [45]. Recently, Preethi also esterified DXI with dextran (Mw 60,000–90,000 Da) in the one pot chemical reaction using N,N-carbonyldiimidazole [46]. This prodrug shows a log P of 5.4 and in artificial intestinal fluid was significantly hydrolyzed (99.53%) by following first-order kinetics with 85.9 min half-life. During the preclinical experiments in the in vivo models on the Wistar rats, DXI-dextran conjugate showed superior analgesic, anti-inflammatory, antipyretic activities in [46].
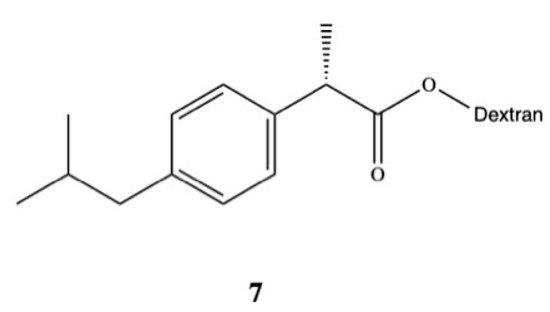
Other groups have also synthetized DXI prodrugs forming DXI-antioxidant conjugates with menthol, sesamol, and umbelliferone 8–10 aimed at reducing gastrointestinal side effects (Figure 6). In order to achieve this purpose, ester analogues of DXI were obtained [47]. The prodrugs were stable in the stomach whereas in plasma they underwent hydrolysis processes releasing DXI. Since minimum hydrolysis was observed at acidic pH (1.2), this may indicate that DXI-ester prodrugs are less irritating that DXI. Moreover, these results were confirmed in vivo performed on male and female mice orally administrated with the synthesized prodrugs [47]. Preclinical experiments showed that these DXI-ester prodrugs caused a significant increase in anti-inflammatory, analgesic, and antipyretic activity [47].
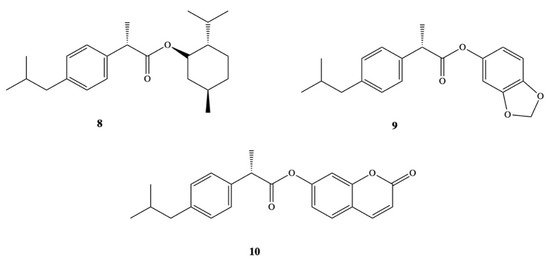
NSAIDs have been reported in the literature as active molecules in the treatment of breast cancer cell line MCF-7 and colon cancer cells HT-29 [48][49][50][51]. Based on these findings, DXI was covalently linked via alkylene spacer units to a riboflavin derivative 11a–e (Figure 7) due to well-known ability of this moiety to reduction of cancer chemotherapy toxicity and protection against oxidant-mediated inflammatory organ injury [52]. Synthesized conjugates of DXI with 2′,3′,4′,5′-tetraacetylriboflavin (TAR) exhibited comparable antiproliferative activity when used in the treatment of chemotherapy 5-fluorouracil (5-FU) towards both tested cancer cell lines MCF-7 and HT-29 with IC50 values in the range of 7.8–14.9 M. The authors have interpreted observed cytotoxicity of synthesized prodrugs by intracellular release of DXI or by the fact that conjugates are able to act not only as drug delivery agents but independent active molecules [53].
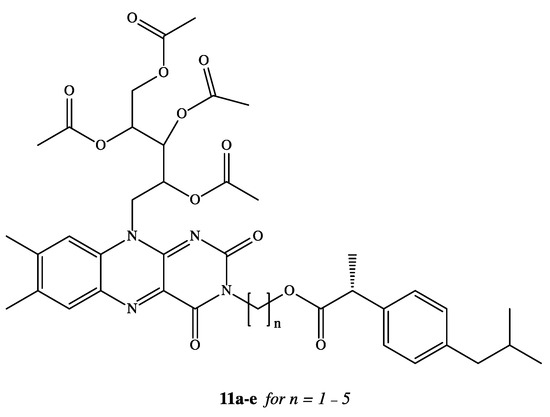
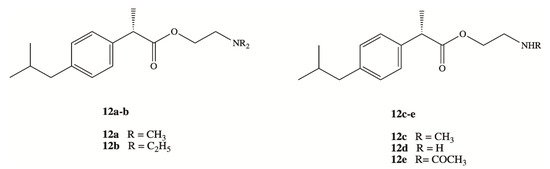
Clinical application of DXI and other NSAIDs in the treatment of neurodegenerative disorders is strongly limited by the lack of their brain accessibility. In order to overcome limited distribution of DXI in the central nervous system (CNS), Zhang et al. have designed and synthesized five prodrugs 12a–e (Figure 8) [54]. These compounds were obtained via esterification reaction of the carboxy group of DXI with ethanolamine with substituted amino group by methyl, ethyl and acetyl groups and their brain-targeting efficiency was evaluated on male Sprague-Dawley rats. In the biodistribution study, the brain-to-plasma concentration ratios were 9.31- to 17.0-fold higher for DXI derivatives modified by ethanolamine related structures than for DXI after 10 min from their intravenous administration. The mechanism responsible for this enhancement was next studied by Li et al. using an in vitro blood brain barrier (BBB) model and in situ perfusion technique [55]. DXI prodrugs with a primary and secondary amine enter via passive diffusion, whereas prodrugs with tertiary amine modifications seem to enter using an active process that is energy and pH dependent but was independent of sodium or membrane potential [55]. Since the experiments were carried out in vitro, DXI prodrug with a tertiary amine could constitute a potential suitable approximation in order to increase transport across the BBB using the pyrilamine-sensitive H+/OC antiporter. However, in vivo results will be necessary to confirm its efficacy [55].
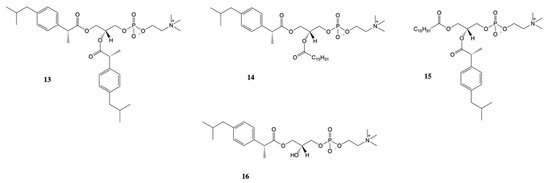
New approach for the synthesis of prodrugs of DXI was presented by Kłobucki and coworkers [56]. They obtained a series of novel phosphatidylcholines 13–16 containing dexibuprofen in their structures (Figure 9), which were subsequently evaluated towards two cancer cells (HL-60 and Caco-2) and normal iporcine epithelial intestinal (PEC-J2) cells. Lysophosphatidylcholine containing DXI in the sn-1 position 16 and asymmetrically substituted phosphatidylcholine 14–15 were characterized by lower toxicity than DXI against HL-60 cells. The obtained results confirmed presented in the literature data indicate that phospholipid derivatives of active compounds are less cytotoxic than the compounds themselves and that phospholipids could be used also as an effective carriers of NSAIDs [57][58].
However, despite DXI prodrugs attempts of some authors, a high number of modifications have not been attempted with this compound. This opens a window for future research of more effective and less damaging DXI prodrugs able to undergo clinical practice.
References
- Wu, D.; Bai, X.; Lee, P.; Yang, Y.; Windsor, J.; Qian, J. A systematic review of NSAIDs treatment for acute pancreatitis in animal studies and clinical trials. Clin. Res. Hepatol. Gastroenterol. 2020, 1, 1–18.
- Fosslien, E. Adverse effects of nonsteroidal anti-inflammatory drugs on the gastrointestinal system. Ann. Clin. Lab. Sci. 1998, 28, 67–81.
- Brater, D.C. Clinical Pharmacology of NSAIDs. Clin. Pharmacol. 1988, 128, 1121–1132.
- Rainsford, K.D. Ibuprofen: Pharmacology, efficacy and safety. Inflammopharmacology 2009, 17, 275–342.
- Kaehler, S.T.; Phleps, W.; Hesse, E. Dexibuprofen: Pharmacology, therapeutic uses and safety. Inflammopharmacology 2003, 11, 371–383.
- Chantaburanan, T.; Teeranachaideekul, V.; Chantasart, D.; Jintapattanakit, A.; Junyaprasert, V.B. Effect of binary solid lipid matrix of wax and triglyceride on lipid crystallinity, drug-lipid interaction and drug release of ibuprofen-loaded solid lipid nanoparticles (SLN) for dermal delivery. J. Colloid Interface Sci. 2017, 504, 247–256.
- Hanif, A.M.; Sial, A.A.; Ali, H.; Zafar, F.; Baig, M.T.; Bushra, R.; Khan, M.A.; Nawab, A.; Mustapha, O.; Shafique, S. Dexibuprofen: Statistical assessment of drug release kinetics and investigative quality perspective. Pak. J. Pharm. Sci. 2018, 31, 2157–2162.
- Phleps, W. Overview on clinical data of dexibuprofen. Clin. Rheumatol. 2001, 20, S15–S21.
- Khalid, Q.; Ahmad, M.; Usman, M.; Batool, F.; Shamshad, N.; Rehman, M. Novel β -cyclodextrin nanosponges by chain growth condensation for solubility enhancement of dexibuprofen: Characterization and acute oral toxicity studies. J. Drug Deliv. Sci. Technol. 2021, 61, 102089.
- Evans, A.M.; Nation, R.L.; Sansom, L.N.; Bochner, F.; Somogyi, A.A. Effect of racemic ibuprofen dose on the magnitude and duration of platelet cyclo-oxygenase inhibition: Relationship between inhibition of thromboxane production and the plasma unbound concentration of S(+)-ibuprofen. Br. J. Clin. Pharmacol. 1991, 31, 131–138.
- Wsól, V.; Skálová, L.; Szotáková, B. Chiral inversion of drugs: Coincidence or principle? Curr. Drug Metab. 2004, 5, 517–533.
- Yoon, J.S.; Jeong, D.C.; Oh, J.W.; Lee, K.Y.; Lee, H.S.; Koh, Y.Y.; Kim, J.T.; Kang, J.H.; Lee, J.S. The effects and safety of dexibuprofen compared with ibuprofen in febrile children caused by upper respiratory tract infection. Br. J. Clin. Pharmacol. 2008, 66, 854–860.
- Laska, E.M.; Sunshine, A.; Marrero, I.; Olson, N.; Siegel, C.; McCormick, N. The correlation between blood levels of ibuprofen and clinical analgesic response. Clin. Pharmacol. Ther. 1986, 40, 1–7.
- Rudy, A.C.; Knight, M.P.; Craig Brater, D.; Hall, S.D. Stereoselective administration metabolism of Ibuprofen in humans: Administration of R-, S- and Racemic lbuprofen. J. Pharmacol. Exp. Ther. 1991, 259, 1133–1139.
- Brocks, D.; Jamali, F. The pharmacokinetics of ibuprofen in humans and animals. In Ibuprofen. A Critical Bibliopgraphic Review; Rainsford, K.D., Ed.; Taylor & Francis: London, UK, 1999; pp. 89–142.
- Jeffrey, P.; Tucker, G.T.; Bye, A.; Crewe, H.K.; Wright, P.A. The Site of Inversion of R (–) -Ibuprofen: Studies Using Rat In-situ Isolated Perfused Intestine/liver Preparations. J. Pharm. Pharmacol 1991, 43, 715–720.
- Jamali, F.; Mehvar, R.; Russell, A.S.; Sattari, S.; Yakimets, W.W.; Koo, J. Human Pharmacokinetics of Ibuprofen Enantiomers following Different Doses and Formulations: Intestinal Chiral Inversion. Am. Pharm. Assoc. J. 1992, 81, 221–225.
- Rudy, A.C.; Bradley, J.D.; Ryan, S.I.; Kalasinski, L.A.; Xiaotao, Q.; Hall, S.D. Variability in the disposition of ibuprofen enantiomers in osteoarthritis patients. Ther. Drug Monit. 1992, 14, 464–470.
- Jamali, F.; Kunz-dober, C.M. Pain-mediated altered absorption and metabolism of ibuprofen: An explanation for decreased serum enantiomer concentration after dental surgery. Br. J. Clin. Pharmacol 1999, 47, 391–396.
- Woodman, T.J.; Wood, P.J.; Thompson, A.S.; Hutchings, T.J.; Steel, G.R.; Jiao, P.; Threadgill, M.D.; Lloyd, M.D. Chiral inversion of 2-arylpropionyl-CoA esters by human a-methylacyl-CoA racemase 1A (P504S)—A potential mechanism for the anti-cancer effects of ibuprofen. Chem. Commun. 2011, 47, 7332–7334.
- Davies, N.M. Clinical Pharmacokinetics of Ibuprofen. The First 30 Years. Clin. Pharmacokinet. 1998, 34, 101–154.
- Lloyd, M.D.; Yevglevskis, M.; Lee, G.L.; Wood, P.J.; Threadgill, M.D.; Woodman, T.J. Progress in Lipid Research a -Methylacyl-CoA racemase (AMACR): Metabolic enzyme, drug metabolizer and cancer marker P504S. Prog. Lipid Res. 2013, 52, 220–230.
- Bonabello, A.; Galmozzi, M.R.; Canaparo, R.; Isaia, G.C.; Serpe, L.; Muntoni, E.; Zara, G.P. Dexibuprofen (S(+)-isomer ibuprofen) reduces gastric damage and improves analgesic and antiinflammatory effects in rodents. Anesth. Analg. 2003, 97, 402–408.
- Hawel, R.; Klein, G.; Singer, F.; Mayrhofer, F.; Kähler, S.T. Comparison of the efficacy and tolerability of dexibuprofen and celecoxib in the treatment of osteoarthritis of the hip. Int. J. Clin. Pharmacol. Ther. 2003, 41, 153–164.
- Repetti, M.R.; Benedetich, C.; Jos, C.; Briand, L.E.; Cornaglia, L.M.; Bosko, M.L. Isolation of ibuprofen enantiomers and racemic esters through electrodialysis. J. Membr. Sci. 2021, 618, 1187714.
- Leising, G.; Resel, R.; Stelzer, F.; Lanziner, A.; Hantich, G.; Tasch, S. Physical aspects of dexibuprofen and racemic ibuprofen. J. Clin. Pharmacol. 1996, 36, 2–6.
- Derry, S.; Best, J.; Moore, R.A. Single dose oral dexibuprofen [S(+)-ibuprofen] for acute postoperative pain in adults. Cochrane Database Syst. Rev. 2013, 2017.
- Apsen Farmaceutica S.A. Phase I Study to Evaluate the Safety of Dexibuprofen 300 mg Under Fasting and Fed Conditions. Available online: (accessed on 18 November 2020).
- Apsen Farmaceutica S.A. Phase I Study to Evaluate the Safety of Dexibuprofen 200 mg Under Fasting and Fed Conditions. Available online: (accessed on 14 December 2017).
- University, Inje. The Efficacy and Safety of Dexibuprofen Syrup. Available online: (accessed on 18 January 2010).
- Gebro Pharma GmbH. Dexibuprofen 400 mg Sachet Versus Ibuprofen 400 mg Sachet in Patients With Osteoarthritis of the Hip or Knee. Available online: (accessed on 10 July 2012).
- Gebro Pharma GmbH. Dexibuprofen Powder for Oral Suspension. Available online: (accessed on 18 November 2020).
- Koziolek, M.; Grimm, M.; Schneider, F.; Jedamzik, P.; Sager, M.; Kühn, J.P.; Siegmund, W.; Weitschies, W. Navigating the human gastrointestinal tract for oral drug delivery: Uncharted waters and new frontiers. Adv. Drug Deliv. Rev. 2016, 101, 75–88.
- Keep, E.R.; Sidelmann, U.G.; Hansen, S.H. Isolation and characteritztion of major phase I and II metabolites of Ibuprofen. Pharm. Res. 1997, 14, 676–680.
- Hao, H.; Wang, G.; Sun, J. Enantioselective Pharmacokinetics of Ibuprofen and Involved Mechanisms. Drug Metab. Rev. 2005, 1, 215–234.
- Schneider, H.T.; Nuernberg, B.; Dietzel, K.; Brune, K.A.Y. Biliary elimination of non-steroidal anti-inflammatory drugs in patients. Br. J. Clin. Pharmacol. 1990, 29, 127–131.
- Hamman, M.A.; Thompson, G.A.; Hall, S.D. Regioselective and Stereoselective Metabolism of Ibuprofen by Human Cytochrome I? 450 2C. Biochem. Pharmacol. 1997, 54, 33–41.
- Walker, J.S.; Carmody, J.J. Experimental Pain in Healthy Human Subjects: Gender Differences in Nociception and in Response to Ibuprofen. Anesth. Analg. 1998, 86, 157–1262.
- Peesa, J.P.; Yalavarthi, P.R.; Rasheed, A.; Mandava, V.B.R. A perspective review on role of novel NSAID prodrugs in the management of acute inflammation. J. Acute Dis. 2016, 5, 364–381.
- Qandil, A.M. Prodrugs of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), More Than Meets the Eye: A Critical Review. Int. J. Mol. Sci. 2012, 13, 7244.
- Ashraf, Z.; Imram, M.; Amin, S. Synthesis, characteritzation and in vitro hydrolysis studies of ester and amide prodrugs of dexibuprofen. Med. Chem. Res. 2012, 21, 3361–3368.
- Ashraf, Z.; Mahmood, T.; Hassan, M.; Afzal, S.; Rafique, H.; Afzal, K.; Latip, J. Dexibuprofen amide derivatives as potential anticancer agents: Synthesis, in silico docking, bioevaluation, and molecular dynamic simulation. Drug Des. Dev. Ther. 2019, 13, 1643–1657.
- Arshad, N.; Zafran, M.; Ashraf, Z.; Perveen, F. Synthesis, characterization of amide substituted dexibuprofen derivatives and their spectral, voltammetric and docking investigations for DNA binding interactions. J. Photochem. Photobiol. B Biol. 2017, 169, 134–147.
- Rasheed, A.; Kumar, C.K.A.; Mishra, A.; Rasheed, A.; Kumar, C.K.A.; Mishra, A. Synthesis, hydrolysis studies and phamacodynamic profiles of amide prodrugs of dexibuprofen with amino acids Synthesis, hydrolysis studies and phamacodynamic profiles of amide prodrugs of dexibuprofen with amino acids. J. Enzyme Inhib. Med. Chem. 2011, 26, 688–695.
- Parmar, S.; Amit Gangwal, N.S.D. Dexibuprofen-Dextran macromolecular prodrugs: Synthesis, characterization and pharmacological evaluation. Sch. Libr. Res. 2011, 2, 373–383.
- JayaPreethu, P.; Lakshmanarao, A.; Basaveswara, M.V. Design and Schematic Evaluation of Dextran Conjugated Dexibuprofen, a Gastrosparing NSAID. Am. J. Chem. 2018, 3, 30–41.
- Ashraf, Z.; Alamgeer; Rasool, R.; Hassan, M.; Ahsan, H.; Afzal, S.; Afzal, K.; Cho, H.; Kim, S.J. Synthesis, bioevaluation and molecular dynamic simulation studies of dexibuprofen-antioxidant mutual prodrugs. Int. J. Mol. Sci. 2016, 17, 2151.
- Mazhar, D.; Ang, R.; Waxman, J. COX inhibitors and breast cancer. Br. J. Cancer 2006, 346–350.
- Giardello, F.; Offerhaus, G.J.A.; DuBois, R.N. The role of nonsteroidal anti-inflammatory drugs in colorectal cancer prevention. Eur. J. Cancer 1995, 31A, 1071.
- Cai, Y.; Yousef, A.; Grandis, J.R.; Johnson, D.E. NSAID therapy for PIK3CA-Altered colorectal, breast, and head and neck cancer. Adv. Biol. Regul. 2020, 75, 1000653.
- Marshall, S.F.; Bernstein, L.; Anton-Culver, H.; Deapen, D.; Horn-Ross, P.L.; Mohrenweiser, H.; Pell, D.; Pinder, R.; Purdie, D.M.; Reynolds, P.; et al. Nonsteroidal anti-inflammatory drug use and breast cancer by stage and hormone receptor. J. Natl. Cancer Inst. 2005, 97, 805–812.
- Seekamp, A.; Hultquist, D.E.; Till, G.O. Protection By Vitamin B 2 Against Oxidant-Mediated Acute Lung Injury. Inflammation 1999, 23, 449–460.
- Banekovich, C.; Ott, I.; Koch, T.; Matuszczak, B.; Gust, R. Synthesis and biological activities of novel dexibuprofen tetraacetylriboflavin conjugates. Bioorg. Med. Chem. Lett. 2007, 17, 683–687.
- Zhang, X.; Liu, X.; Gong, T.; Sun, X.; Zhang, Z. In vitro and in vivo investigation of dexibuprofen derivatives for CNS delivery. Acta Pharmacol. Sin. 2012, 33, 279–288.
- Li, Y.; Zhou, Y.; Jiang, J.; Wang, X.; Fu, Y.; Gong, T.; Sun, X.; Zhang, Z. Mechanism of brain targeting by dexibuprofen prodrugs modified with ethanolamine-related structures. J. Cereb. Blood Flow Metab. 2015, 35, 1985–1994.
- Kłobucki, M.; Urbaniak, A.; Grudniewska, A.; Kocbach, B.; Maciejewska, G.; Kiełbowicz, G.; Czesław, M.; Wawrzeńczyk, U. Syntheses and cytotoxicity of phosphatidylcholines containing ibuprofen or naproxen moieties. Sci. Rep. 2019, 9, 220.
- Gliszczyńska, A.; Niezgoda, N.; Gladkowski, W.; Świtalska, M.; Wietrzyk, J. Isoprenoid-phospholipid conjugates as potential therapeutic agents: Synthesis, characterization and antiproliferative studies. PLoS ONE 2017, 12, e0172238.
- Gliszczyńska, A.; Niezgoda, N.; Gładkowski, W.; Czarnecka, M.; Świtalska, M.; Wietrzyk, J. Synthesis and biological evaluation of novel phosphatidylcholine analogues containing monoterpene acids as potent antiproliferative agents. PLoS ONE 2016, 11, e0157278.


