 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Olga Lavrik | + 4002 word(s) | 4002 | 2021-06-15 08:46:21 | | | |
| 2 | Olga Lavrik | Meta information modification | 4002 | 2021-06-20 19:34:09 | | | | |
| 3 | Vivi Li | Meta information modification | 4002 | 2021-06-21 04:41:51 | | | | |
| 4 | Conner Chen | Meta information modification | 4002 | 2021-10-12 05:16:18 | | | | |
| 5 | Conner Chen | Meta information modification | 4002 | 2021-10-12 05:16:36 | | |
Video Upload Options
Nucleotide excision repair (NER) is the most versatile DNA repair pathway, which can remove diverse bulky DNA lesions destabilizing a DNA duplex. NER substrates are UV photoproducts, e.g., cyclobutane pyrimidine dimers (CPDs), pyrimidine-pyrimidone-(6-4)-photoproducts (6-4PPs), intrastrand crosslinks, and bulky adducts of DNA bases with reactive metabolites of some chemical carcinogens or chemotherapeutic agents. These kinds of lesions can be substrates for two NER sub-pathways—global genome NER (GG-NER) and transcription-coupled NER (TC-NER)—that overlap, except for the mode of DNA damage recognition.
NER defects cause several autosomal recessive genetic disorders. Xeroderma pigmentosum (XP) is one of the NER-associated syndromes characterized by low efficiency of the removal of bulky DNA adducts generated by ultraviolet radiation. XP patients have extremely high ultraviolet-light sensitivity of sun-exposed tissues, often resulting in multiple skin and eye cancers.
1. Introduction
2. Nucleotide Excision Repair
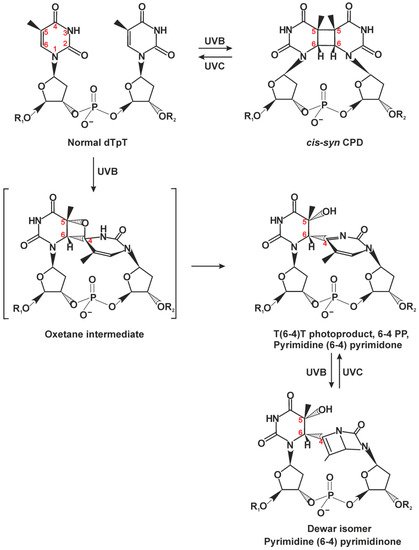
2.1. Classic NER Substrates
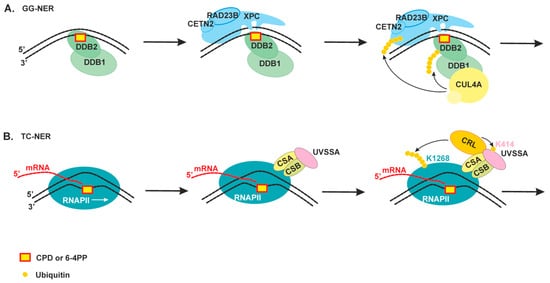
2.2. The Damage Recognition Step
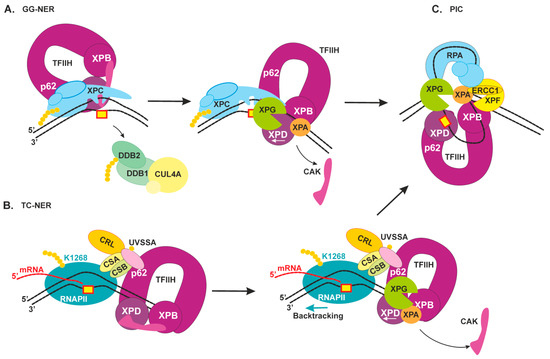
2.3. Damage Verification and Pre-Incision Complex Formation
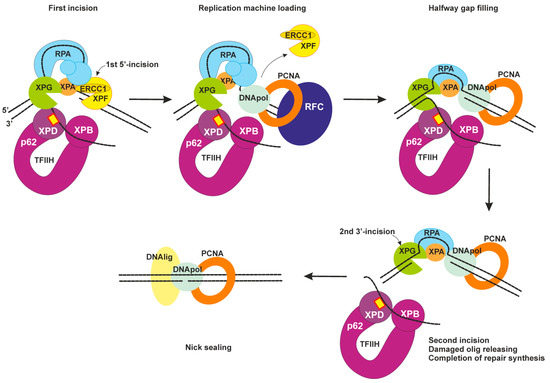
2.4. Dual Incision, Resynthesis, and Ligation
References
- Kumar, N.; Moreno, N.C.; Feltes, B.C.; Menck, C.F.; Houten, B.V. Cooperation and interplay between base and nucleotide excision repair pathways: From DNA lesions to proteins. Genet Mol. Biol. 2020, 43 (Suppl. 1), e20190104.
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200.
- Kumar, N.; Raja, S.; Van Houten, B. The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic Acids Res. 2020, 48, 11227–11243.
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583.
- Limpose, K.L.; Corbett, A.H.; Doetsch, P.W. BERing the burden of damage: Pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair (Amst.) 2017, 56, 51–64.
- Kusakabe, M.; Onishi, Y.; Tada, H.; Kurihara, F.; Kusao, K.; Furukawa, M.; Iwai, S.; Yokoi, M.; Sakai, W.; Sugasawa, K. Mechanism and regulation of DNA damage recognition in nucleotide excision repair. Genes Environ. 2019, 41, 2.
- Mu, H.; Geacintov, N.E.; Broyde, S.; Yeo, J.E.; Schärer, O.D. Molecular basis for damage recognition and verification by XPC-RAD23B and TFIIH in nucleotide excision repair. DNA Repair (Amst.) 2018, 71, 33–42.
- Gillet, L.C.; Scharer, O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006, 106, 253–276.
- Son, K.; Schärer, O.D. Repair, Removal, and Shutdown: It All Hinges on RNA Polymerase II Ubiquitylation. Cell 2020, 180, 1039–1041.
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970.
- Rapin, I.; Lindenbaum, Y.; Dickson, D.W.; Kraemer, K.H.; Robbins, J.H. Cockayne syndrome and xeroderma pigmentosum. Neurology 2000, 55, 1442–1449.
- DiGiovanna, J.J.; Kraemer, K.H. Shining a light on xeroderma pigmentosum. J. Investig. Dermatol. 2012, 132, 785–796.
- Fassihi, H.; Sethi, M.; Fawcett, H.; Wing, J.; Chandler, N.; Mohammed, S.; Craythorne, E.; Morley, A.M.; Lim, R.; Turner, S.; et al. Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc. Natl. Acad. Sci. USA 2016, 113, E1236–E1245.
- Bradford, P.T.; Goldstein, A.M.; Tamura, D.; Khan, S.G.; Ueda, T.; Boyle, J.; Oh, K.S.; Imoto, K.; Inui, H.; Moriwaki, S.; et al. Cancer and neurologic degeneration in xeroderma pigmentosum: Long term follow-up characterises the role of DNA repair. J. Med. Genet. 2011, 48, 168–176.
- Brooks, P.J. The Case for 8, 5′-Cyclopurine-2′-Deoxynucleosides as Endogenous DNA Lesions That Cause Neurodegeneration in Xeroderma Pigmentosum. Neuroscience 2007, 145, 1407–1417.
- Okur, M.N.; Fang, E.F.; Fivenson, E.M.; Tiwari, V.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome proteins CSA and CSB maintain mitochondrial homeostasis through NAD+ signaling. Aging Cell 2020, 19, e13268.
- Tiwari, V.; Baptiste, B.A.; Okur, M.N.; Bohr, V.A. Current and emerging roles of Cockayne syndrome group B (CSB) protein. Nucleic Acids Res. 2021, 49, 2418–2434.
- Saki, M.; Prakash, A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic. Biol. Med. 2017, 107, 216–227.
- Roca-Portoles, A.; Tait, S.W.G. Mitochondrial quality control: From molecule to organelle. Cell Mol. Life Sci. 2021. Online ahead of print.
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Bohr, V.A. Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders. Autophagy 2014, 10, 1468–1469.
- Patel, J.; Baptiste, B.A.; Kim, E.; Hussain, M.; Croteau, D.L.; Bohr, V.A. DNA damage and mitochondria in cancer and aging. Carcinogenesis 2020, 41, 1625–1634.
- Yokoyama, H.; Mizutani, R. Structural biology of DNA (6-4) photoproducts formed by ultraviolet radiation and interactions with their binding proteins. Int. J. Mol. Sci. 2014, 15, 20321–20338.
- Schärer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609.
- Kim, J.K.; Patel, D.; Choi, B.S. Contrasting structural impacts induced by cis-syn cyclobutane dimer and (6-4) adduct in DNA duplex decamers: Implication in mutagenesis and repair activity. Photochem. Photobiol. 1995, 62, 44–50.
- Reardon, J.T.; Sancar, A. Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 2003, 17, 2539–2551.
- Krasikova, Y.S.; Rechkunova, N.I.; Maltseva, E.A.; Craescu, C.T.; Petruseva, I.O.; Lavrik, O.I. Influence of centrin 2 on the interaction of nucleotide excision repair factors with damaged DNA. Biochemistry 2012, 77, 346–353.
- Min, J.H.; Pavletich, N.P. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 2007, 449, 570–575.
- Paul, D.; Mu, H.; Zhao, H.; Ouerfelli, O.; Jeffrey, P.D.; Broyde, S.; Min, J.H. Structure and mechanism of pyrimidine-pyrimidone (6-4) photoproduct recognition by the Rad4/XPC nucleotide excision repair complex. Nucleic Acids Res. 2019, 47, 6015–6028.
- Cheon, N.Y.; Kim, H.S.; Yeo, J.E.; Scharer, O.D.; Lee, J.Y. Single-molecule visualization reveals the damage search mechanism for the human NER protein XPC-RAD23B. Nucleic Acids Res. 2019, 47, 8337–8347.
- Paul, D.; Mu, H.; Tavakoli, A.; Dai, Q.; Chen, X.; Chakraborty, S.; He, C.; Ansari, A.; Broyde, S.; Min, J.H. Tethering-facilitated DNA’ opening’ and complementary roles of β-hairpin motifs in the Rad4/XPC DNA damage sensor protein. Nucleic Acids Res. 2020, 48, 12348–12364.
- Buterin, T.; Meyer, C.; Giese, B.; Naegeli, H. DNA quality control by conformational readout on the undamaged strand of the double helix. Chem. Biol. 2005, 12, 913–922.
- Lukyanchikova, N.V.; Petruseva, I.O.; Evdokimov, A.N.; Koroleva, L.S.; Lavrik, O.I. DNA Bearing Bulky Fluorescent and Photoreactive Damage in Both Strands as Substrates of the Nucleotide Excision Repair System. Mol. Biol. (Mosk.) 2018, 52, 277–288.
- Ribeiro-Silva, C.; Sabatella, M.; Helfricht, A.; Marteijn, J.A.; Theil, A.F.; Vermeulen, W.; Lans, H. Ubiquitin and TFIIH-stimulated DDB2 dissociation drives DNA damage handover in nucleotide excision repair. Nat. Commun. 2020, 11, 4868.
- Sugasawa, K.; Okuda, Y.; Saijo, M.; Nishi, R.; Matsuda, N.; Chu, G.; Mori, T.; Iwai, S.; Tanaka, K.; Tanaka, K.; et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 2005, 121, 387–400.
- Nakazawa, Y.; Hara, Y.; Oka, Y.; Komine, O.; van den Heuvel, D.; Guo, C.; Daigaku, Y.; Isono, M.; He, Y.; Shimada, M.; et al. Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair. Cell 2020, 180, 1228–1244.e24.
- Rechkunova, N.I.; Maltseva, E.A.; Lavrik, O.I. Post-translational Modifications of Nucleotide Excision Repair Proteins and Their Role in the DNA Repair. Biochemistry 2019, 84, 1008–1020.
- van der Weegen, Y.; Golan-Berman, H.; Mevissen, T.E.T.; Apelt, K.; González-Prieto, R.; Goedhart, J.; Heilbrun, E.E.; Vertegaal, A.C.O.; van den Heuvel, D.; Walter, J.C.; et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 2020, 11, 2104.
- Tufegdžić Vidaković, A.; Mitter, R.; Kelly, G.P.; Neumann, M.; Harreman, M.; Rodríguez-Martínez, M.; Herlihy, A.; Weems, J.C.; Boeing, S.; Encheva, V.; et al. Regulation of the RNAPII Pool Is Integral to the DNA Damage Response. Cell 2020, 180, 1245–1261.
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411.
- Shivji, M.K.K.; Renaudin, X.; Williams, Ç.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039.
- Tsutakawa, S.E.; Tsai, C.L.; Yan, C.; Bralić, A.; Chazin, W.J.; Hamdan, S.M.; Schärer, O.D.; Ivanov, I.; Tainer, J.A. Envisioning how the prototypic molecular machine TFIIH functions in transcription initiation and DNA repair. DNA Repair (Amst.) 2020, 96, 102972.
- Schilbach, S.; Hantsche, M.; Tegunov, D.; Dienemann, C.; Wigge, C.; Urlaub, H.; Cramer, P. Structures of transcription pre-initiation complex with TFIIH and Mediator. Nature 2017, 551, 204–209.
- Greber, B.J.; Toso, D.B.; Fang, J.; Nogales, E. The complete structure of the human TFIIH core complex. eLife 2019, 8, e44771.
- Kokic, G.; Chernev, A.; Tegunov, D.; Dienemann, C.; Urlaub, H.; Cramer, P. Structural basis of TFIIH activation for nucleotide excision repair. Nat. Commun. 2019, 10, 2885.
- He, Y.; Yan, C.; Fang, J.; Inouye, C.; Tjian, R.; Ivanov, I.; Nogales, E. Near-atomic resolution visualization of human transcription promoter opening. Nature 2016, 533, 359–365.
- Li, C.L.; Golebiowski, F.M.; Onishi, Y.; Samara, N.L.; Sugasawa, K.; Yang, W. Tripartite DNA Lesion Recognition and Verification by XPC, TFIIH, and XPA in Nucleotide Excision Repair. Mol. Cell 2015, 59, 1025–1034.
- Krasikova, Y.S.; Rechkunova, N.I.; Maltseva, E.A.; Petruseva, I.O.; Lavrik, O.I. Localization of xeroderma pigmentosum group A protein and replication protein A on damaged DNA in nucleotide excision repair. Nucleic Acids Res. 2010, 38, 8083–8094.
- Sugitani, N.; Sivley, R.M.; Perry, K.E.; Capra, J.A.; Chazin, W.J. XPA: A key scaffold for human nucleotide excision repair. DNA Repair (Amst.) 2016, 44, 123–135.
- Beckwitt, E.C.; Jang, S.; Carnaval Detweiler, I.; Kuper, J.; Sauer, F.; Simon, N.; Bretzler, J.; Watkins, S.C.; Carell, T.; Kisker, C.; et al. Single molecule analysis reveals monomeric XPA bends DNA and undergoes episodic linear diffusion during damage search. Nat. Commun. 2020, 11, 1356.
- Chen, R.; Wold, M.S. Replication protein A: Single-stranded DNA’s first responder: Dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 2014, 36, 1156–1161.
- Krasikova, Y.S.; Rechkunova, N.I.; Maltseva, E.A.; Lavrik, O.I. RPA and XPA interaction with DNA structures mimicking intermediates of the late stages in nucleotide excision repair. PLoS ONE 2018, 13, e0190782.
- Topolska-Woś, A.M.; Sugitani, N.; Cordoba, J.J.; Le Meur, K.V.; Le Meur, R.A.; Kim, H.S.; Yeo, J.E.; Rosenberg, D.; Hammel, M.; Schärer, O.D.; et al. A key interaction with RPA orients XPA in NER complexes. Nucleic Acids Res. 2020, 48, 2173–2188.
- Fan, J.; Pavletich, N.P. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012, 26, 2337–2347.
- Tsodikov, O.V.; Ivanov, D.; Orelli, B.; Staresincic, L.; Shoshani, I.; Oberman, R.; Schärer, O.D.; Wagner, G.; Ellenberger, T. Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 2007, 26, 4768–4776.
- Fagbemi, A.F.; Orelli, B.; Schärer, O.D. Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair (Amst.) 2011, 10, 722–729.
- Staresincic, L.; Fagbemi, A.F.; Enzlin, J.H.; Gourdin, A.M.; Wijgers, N.; Dunand-Sauthier, I.; Giglia-Mari, G.; Clarkson, S.G.; Vermeulen, W.; Schärer, O.D. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009, 28, 1111–1120.
- Overmeer, R.M.; Moser, J.; Volker, M.; Kool, H.; Tomkinson, A.E.; van Zeeland, A.A.; Mullenders, L.H.; Fousteri, M. Replication protein A safeguards genome integrity by controlling NER incision events. J. Cell Biol. 2011, 192, 401–415.
- Gilljam, K.M.; Müller, R.; Liabakk, N.B.; Otterlei, M. Nucleotide excision repair is associated with the replisome and its efficiency depends on a direct interaction between XPA and PCNA. PLoS ONE 2012, 7, e49199.
- Yu, S.L.; Kang, M.S.; Kim, H.Y.; Gorospe, C.M.; Kim, T.S.; Lee, S.K. The PCNA binding domain of Rad2p plays a role in mutagenesis by modulating the cell cycle in response to DNA damage. DNA Repair (Amst.) 2014, 16, 1–10.
- Kemp, M.G.; Reardon, J.T.; Lindsey-Boltz, L.A.; Sancar, A. Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 2012, 287, 22889–22899.
- Sertic, S.; Mollica, A.; Campus, I.; Roma, S.; Tumini, E.; Aguilera, A.; Muzi-Falconi, M. Coordinated Activity of Y Family TLS Polymerases and EXO1 Protects Non-S Phase Cells from UV-Induced Cytotoxic Lesions. Mol. Cell 2018, 70, 34–47.e4.
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.; Fousteri, M. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol. Cell 2007, 27, 311–323.


