 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michael Kemp | + 2967 word(s) | 2967 | 2021-06-09 06:16:24 | | | |
| 2 | Vivi Li | Meta information modification | 2967 | 2021-06-21 04:32:13 | | |
Video Upload Options
Spironolactone (SP) is commonly used for the treatment of heart failure, hypertension, and complications of cirrhosis by antagonizing the mineralocorticoid receptor. However, SP also antagonizes the androgen receptor, and thus SP has also been shown to be effective in the treatment of acne, hair loss, and hirsutism in women. Interestingly, recent drug repurposing screens have identified new and diverse functions for SP as a simulator of tumor immunosurveillance and as an inhibitor of DNA repair and viral infection. These novel pharmacological effects of SP have all been linked to the ability of SP to induce the rapid proteolytic degradation of the xeroderma pigmentosum group B (XPB) protein. XPB is a critical enzymatic component of the multi-subunit complex known as transcription factor II-H (TFIIH), which plays essential roles in both DNA repair and the initiation of transcription. Given the critical functions for XPB and TFIIH in these processes, the loss of XPB by SP could lead to mutagenesis. However, the ability of SP to promote cancer stem cell death and facilitate immune recognition may counteract the negative consequences of SP to mitigate carcinogenic risk. Thus, SP appears to have new and interesting pharmacological effects that may extend its potential uses.
1. Introduction
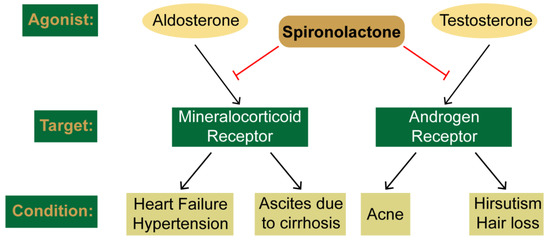
2. Clinical Uses and Canonical Targets of Spironolactone
2.1. SP as a Mineralocorticoid Receptor Antagonist
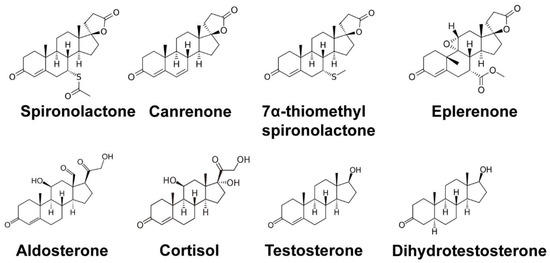
2.2. SP as an Androgen Receptor Antagonist
3. The XPB Protein as a Novel Target of Spironolactone
3.1. Identification of the DNA Repair Protein XPB as a Target of SP
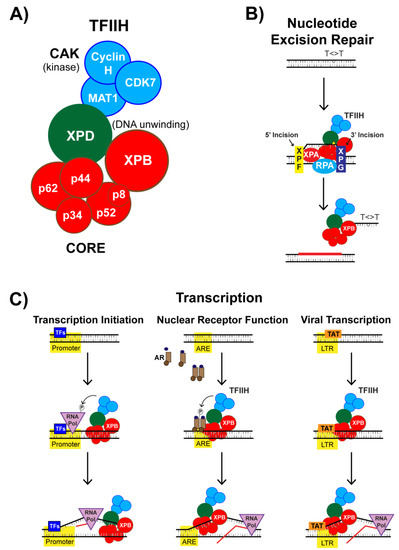
3.2. SP Impacts TFIIH Function in Transcription Initiation
3.3. Nuclear Receptor-Dependent Transcription is Regulated by TFIIH and SP
3.4. SP Impacts TFIIH Function in Viral Transcription
References
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by repurposing: Teaching new tricks to old dogs. Trends Pharmacol. Sci. 2013, 34, 508–517.
- Leung, W.-H.; Vong, Q.P.; Lin, W.; Janke, L.; Chen, T.; Leung, W. Modulation of NKG2D ligand expression and metastasis in tumors by spironolactone via RXRγ activation. J. Exp. Med. 2013, 210, 2675–2692.
- Alekseev, S.; Ayadi, M.; Brino, L.; Egly, J.-M.; Larsen, A.K.; Coin, F. A Small Molecule Screen Identifies an Inhibitor of DNA Repair Inducing the Degradation of TFIIH and the Chemosensitization of Tumor Cells to Platinum. Chem. Biol. 2014, 21, 398–407.
- Shahar, O.D.; Kalousi, A.; Eini, L.; Fisher, B.; Weiss, A.; Darr, J.; Mazina, O.; Bramson, S.; Kupiec, M.; Eden, A.; et al. A high-throughput chemical screen with FDA approved drugs reveals that the antihypertensive drug Spironolactone impairs cancer cell survival by inhibiting homology directed repair. Nucleic Acids Res. 2014, 42, 5689–5701.
- Verma, D.; Thompson, J.; Swaminathan, S. Spironolactone blocks Epstein–Barr virus production by inhibiting EBV SM protein function. Proc. Natl. Acad. Sci. USA 2016, 113, 3609–3614.
- Kagawa, C.M.; Cella, J.A.; Van Arman, C.G. Action of new steroids in blocking effects of aldosterone and desoxycorticosterone on salt. Science 1957, 126, 1015–1016.
- Liddle, G.W. Sodium Diuresis Induced by Steroidal Antagonists of Aldosterone. Science 1957, 126, 1016–1018.
- Cranston, W.; Juel-Jensen, B. The effects of spironolactone and chlorthalidone on arterial pressure. Lancet 1962, 279, 1161–1164.
- Sturtevant, F.M. Antihypertensive Effects of an Aldosterone Antagonist. Science 1958, 127, 1393–1394.
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The Effect of Spironolactone on Morbidity and Mortality in Patients with Severe Heart Failure. N. Engl. J. Med. 1999, 341, 709–717.
- Zannad, F.; McMurray, J.J.; Krum, H.; Van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B. Eplerenone in Patients with Systolic Heart Failure and Mild Symptoms. N. Engl. J. Med. 2011, 364, 11–21.
- Moore, K.; Aithal, G.P. Guidelines on the management of ascites in cirrhosis. Gut 2006, 55, vi1–vi12.
- Dojki, F.K.; Bakris, G. Nonsteroidal mineralocorticoid antagonists in diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 368–374.
- Bowman, J.; Kim, H.; Bustamante, J. Drug-Induced Gynecomastia. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2012, 32, 1123–1140.
- Bonne, C.; Raynaud, J. Mode of spironolactone anti-androgenic action: Inhibition of androstanolone binding to rat prostate androgen receptor. Mol. Cell. Endocrinol. 1974, 2, 59–67.
- Corvol, P.; Michaud, A.; Menard, J.; Freifeld, M.; Mahoudeau, J. Antiandrogenic Effect of Spirolactones: Mechanism of Action. Endocrinology 1975, 97, 52–58.
- Rathnayake, D.; Sinclair, R. Use of spironolactone in dermatology. Skinmed 2011, 8, 328–332.
- Azarchi, S.; Bienenfeld, A.; Lo Sicco, K.; Marchbein, S.; Shapiro, J.; Nagler, A.R. Androgens in women: Hormone-modulating therapies for skin disease. J. Am. Acad. Dermatol. 2019, 80, 1509–1521.
- Zouboulis, C.C. Acne and sebaceous gland function. Clin. Dermatol. 2004, 22, 360–366.
- Park, H.; Skopit, S. Safety Considerations and Monitoring in Patients Treated with Systemic Medications for Acne. Dermatol. Clin. 2016, 34, 185–193.
- Sinclair, R.; Patel, M.; Dawson, T.; Yazdabadi, A.; Yip, L.; Perez, A.; Rufaut, N. Hair loss in women: Medical and cosmetic approaches to increase scalp hair fullness. Br. J. Dermatol. 2011, 165, 12–18.
- Fung, R.; Hellstern-Layefsky, M.; Tastenhoye, C.; Lega, I.C.; Steele, L. Differential Effects of Cyproterone Acetate vs Spironolactone on Serum High-Density Lipoprotein and Prolactin Concentrations in the Hormonal Treatment of Transgender Women. J. Sex. Med. 2016, 13, 1765–1772.
- Gardner, I.H.; Safer, J.D. Progress on the road to better medical care for transgender patients. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 553–558.
- Menard, R.H.; Martin, H.F.; Stripp, B.; Gillette, J.R.; Bartter, F.C. Spironolactone and cytochrome P-450: Impairment of steroid hydroxylation in the adrenal cortex. Life Sci. 1974, 15, 1639–1648.
- Menard, R.H.; Bartter, F.C.; Gillette, J.R. Spironolactone and cytochrome P-450: Impairment of steroid 21-hydroxylation in the adrenal cortex. Arch. Biochem. Biophys. 1976, 173, 395–402.
- Sancar, A. Mechanisms of DNA Repair by Photolyase and Excision Nuclease (Nobel Lecture). Angew. Chem. Int. Ed. 2016, 55, 8502–8527.
- Oh, K.-S.; Khan, S.; Jaspers, N.G.J.; Raams, A.; Ueda, T.; Lehmann, A.; Friedmann, P.S.; Emmert, S.; Gratchev, A.; Lachlan, K.; et al. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): Xeroderma pigmentosum without and with Cockayne syndrome. Hum. Mutat. 2006, 27, 1092–1103.
- Natale, V.; Raquer, H. Xeroderma pigmentosum-Cockayne syndrome complex. Orphanet J. Rare Dis. 2017, 12, 65.
- Compe, E.; Egly, J.-M. Nucleotide Excision Repair and Transcriptional Regulation: TFIIH and Beyond. Annu. Rev. Biochem. 2016, 85, 265–290.
- Compe, E.; Egly, J.-M. TFIIH: When transcription met DNA repair. Nat. Rev. Mol. Cell Biol. 2012, 13, 343–354.
- Rimel, J.K.; Taatjes, D.J. The essential and multifunctional TFIIH complex. Protein Sci. 2018, 27, 1018–1037.
- Hu, J.; Choi, J.-H.; Gaddameedhi, S.; Kemp, M.G.; Reardon, J.T.; Sancar, A. Nucleotide Excision Repair in Human Cells. J. Biol. Chem. 2013, 288, 20918–20926.
- Kemp, M.G.; Reardon, J.T.; Lindsey-Boltz, L.; Sancar, A. Mechanism of Release and Fate of Excised Oligonucleotides during Nucleotide Excision Repair. J. Biol. Chem. 2012, 287, 22889–22899.
- Oksenych, V.; De Jesus, B.B.; Zhovmer, A.; Egly, J.-M.; Coin, F. Molecular insights into the recruitment of TFIIH to sites of DNA damage. EMBO J. 2009, 28, 2971–2980.
- Marteijn, J.A.; Hoeijmakers, J.; Vermeulen, W. Check, Check …Triple Check: Multi-Step DNA Lesion Identification by Nucleotide Excision Repair. Mol. Cell 2015, 59, 885–886.
- Kemp, M.G.; Krishnamurthy, S.; Kent, M.N.; Schumacher, D.L.; Sharma, P.; Excoffon, K.J.; Travers, J.B. Spironolactone Depletes the XPB Protein and Inhibits DNA Damage Responses in UVB-Irradiated Human Skin. J. Investig. Dermatol. 2019, 139, 448–454.
- Hutcherson, R.J.; Kemp, M.G. ATR kinase inhibition sensitizes quiescent human cells to the lethal effects of cisplatin but increases mutagenesis. Mutat. Res. Mol. Mech. Mutagen. 2019, 111678, 111678.
- Shaj, K.; Hutcherson, R.J.; Kemp, M.G. ATR Kinase Activity Limits Mutagenesis and Promotes the Clonogenic Survival of Quiescent Human Keratinocytes Exposed to UVB Radiation. Photochem. Photobiol. 2019, 96, 105–112.
- Szalat, R.; Samur, M.K.; Fulciniti, M.; López, M.; Nanjappa, P.; Cleynen, A.; Wen, K.; Kumar, S.; Perini, T.; Calkins, A.S.; et al. Nucleotide excision repair is a potential therapeutic target in multiple myeloma. Leukemia 2017, 32, 111–119.
- Zhang, M.; Du, W.; Acklin, S.M.; Jin, S.; Xia, F. SIRT2 protects peripheral neurons from cisplatin-induced injury by enhancing nucleotide excision repair. J. Clin. Investig. 2020.
- Savolainen, L.; Cassel, T.; Helleday, T. The XPD subunit of TFIIH is required for transcription-associated but not DNA double-strand break-induced recombination in mammalian cells. Mutagenesis 2010, 25, 623–629.
- Moriel-Carretero, M.; Aguilera, A. A Postincision-Deficient TFIIH Causes Replication Fork Breakage and Uncovers Alternative Rad51- or Pol32-Mediated Restart Mechanisms. Mol. Cell 2010, 37, 690–701.
- Dungrawala, H.; Rose, K.L.; Bhat, K.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 998–1010.
- Sanomachi, T.; Suzuki, S.; Togashi, K.; Sugai, A.; Seino, S.; Okada, M.; Yoshioka, T.; Kitanaka, C.; Yamamoto, M. Spironolactone, a Classic Potassium-Sparing Diuretic, Reduces Survivin Expression and Chemosensitizes Cancer Cells to Non-DNA-Damaging Anticancer Drugs. Cancers 2019, 11, 1550.
- Fishburn, J.; Tomko, E.; Galburt, E.; Hahn, S. Double-stranded DNA translocase activity of transcription factor TFIIH and the mechanism of RNA polymerase II open complex formation. Proc. Natl. Acad. Sci. USA 2015, 112, 3961–3966.
- Schaeffer, L.; Roy, R.; Humbert, S.; Moncollin, V.; Vermeulen, W.; Hoeijmakers, J.; Chambon, P.; Egly, J. DNA repair helicase: A component of BTF2 (TFIIH) basic transcription factor. Science 1993, 260, 58–63.
- Elinoff, J.M.; Chen, L.-Y.; Dougherty, E.J.; Awad, K.S.; Wang, S.; Biancotto, A.; Siddiqui, A.H.; Weir, N.A.; Cai, R.; Sun, J.; et al. Spironolactone-induced degradation of the TFIIH core complex XPB subunit suppresses NF-κB and AP-1 signalling. Cardiovasc. Res. 2018, 114, 65–76.
- Rochette-Egly, C. Nuclear receptors: Integration of multiple signalling pathways through phosphorylation. Cell. Signal. 2003, 15, 355–366.
- Chymkowitch, P.; Le May, N.; Charneau, P.; Compe, E.; Egly, J.-M. The phosphorylation of the androgen receptor by TFIIH directs the ubiquitin/proteasome process. EMBO J. 2010, 30, 468–479.
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190.
- Arya, S.; Guo, C.; Josephs, S.; Wong-Staal, F. Trans-activator gene of human T-lymphotropic virus type III (HTLV-III). Science 1985, 229, 69–73.
- Sodroski, J.; Rosen, C.; Wong-Staal, F.; Salahuddin, S.; Popovic, M.; Arya, S.; Gallo, R.; Haseltine, W. Trans-acting transcriptional regulation of human T-cell leukemia virus type III long terminal repeat. Science 1985, 227, 171–173.
- Berkhout, B.; Silverman, R.H.; Jeang, K.-T. Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell 1989, 59, 273–282.
- Das, A.T.; Harwig, A.; Berkhout, B. The HIV-1 Tat Protein Has a Versatile Role in Activating Viral Transcription. J. Virol. 2011, 85, 9506–9516.
- Cujec, T.P.; Okamoto, H.; Fujinaga, K.; Meyer, J.; Chamberlin, H.; Morgan, D.O.; Peterlin, B.M. The HIV transactivator TAT binds to the CDK-activating kinase and activates the phosphorylation of the carboxy-terminal domain of RNA polymerase II. Genome Res. 1997, 11, 2645–2657.
- García-Martínez, L.F.; Ivanov, D.; Gaynor, R.B. Association of Tat with Purified HIV-1 and HIV-2 Transcription Preinitiation Complexes. J. Biol. Chem. 1997, 272, 6951–6958.
- Yoder, K.; Sarasin, A.; Kraemer, K.H.; McIlhatton, M.; Bushman, F.; Fishel, R. The DNA repair genes XPB and XPD defend cells from retroviral infection. Proc. Natl. Acad. Sci. USA 2006, 103, 4622–4627.
- Yoder, K.; Roddick, W.; Hoellerbauer, P.; Fishel, R. XPB mediated retroviral cDNA degradation coincides with entry to the nucleus. Virology 2010, 410, 291–298.
- Espeseth, A.S.; Fishel, R.; Hazuda, D.; Huang, Q.; Xu, M.; Yoder, K.E.; Zhou, H. siRNA Screening of a Targeted Library of DNA Repair Factors in HIV Infection Reveals a Role for Base Excision Repair in HIV Integration. PLoS ONE 2011, 6, e17612.
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of Host Proteins Required for HIV Infection Through a Functional Genomic Screen. Science 2008, 319, 921–926.
- Lacombe, B.; Morel, M.; Margottin-Goguet, F.; Ramirez, B.C. Specific Inhibition of HIV Infection by the Action of Spironolactone in T Cells. J. Virol. 2016, 90, 10972–10980.
- Martella, C.; Tollenaere, A.I.; Waast, L.; Lacombe, B.; Groussaud, D.; Margottin-Goguet, F.; Ramirez, B.C.; Pique, C. HTLV-1 Transactivator Tax Exploits The Xpb Subunit Of Tfiih During Viral Transcription. J. Virol. 2020, 8, 02171-19.



