 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natália Cruz-Martins | + 3494 word(s) | 3494 | 2021-06-09 06:01:37 | | | |
| 2 | Peter Tang | Meta information modification | 3494 | 2021-06-17 05:02:14 | | |
Video Upload Options
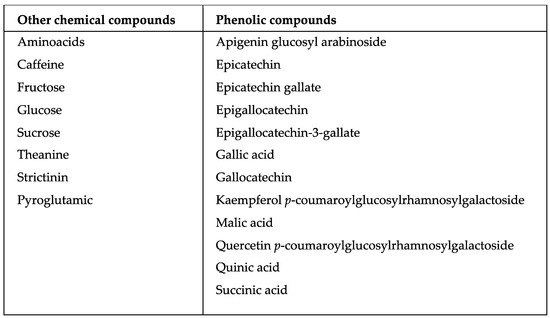
Camellia sinensis is the most consumed beverage worldwide. It contains a wide variety of secondary metabolites, such as alkaloids, saponins, tannins, catechins, and polyphenols, generated through a condensation reaction of cinnamic acid with three malonyl-CoA groups. In addition to the metabolic processes occurring within this plant, there are also some plant-associated bacterial endophytes. These bacteria reside in the living tissues of the host plants without causing any harmful effect to them, thereby stimulating secondary metabolite production with a diverse range of biological effects.
1. Introduction
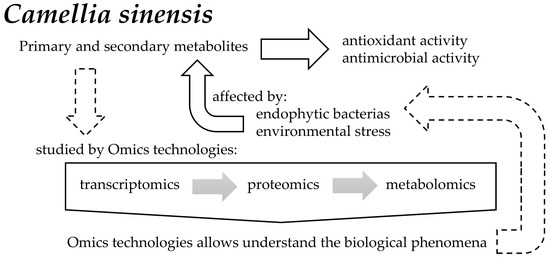
2. Transcriptomics
3. Proteomics
4. Metabolomics
- (1) Target analysis;
- (2) Metabolite profiling;
- (3) Metabolomics; and
- (4) Metabolic fingerprinting [29].
5. Role of Endophytic Bacteria in Camellia sinensis Metabolism
|
Role |
References |
|---|---|
|
Increase defense-related enzymes |
|
|
Increase nitrogen fixation |
|
|
Increase polyphenolic amount |
|
|
Indole-3-acetic acid secretion |
|
|
Inhibition of pathogen growth |
|
|
Phosphate solubilizers |
|
|
Positive plant growth |
|
|
Siderophore producers |
6. Antioxidant and Antimicrobial Properties of Camellia sinensis
References
- Carloni, P.; Tiano, L.; Padella, L.; Bacchetti, T.; Customu, C.; Kay, A.; Damiani, E. Antioxidant activity of white, green and black tea obtained from the same tea cultivar. Food Res. Int. 2013, 53, 900–908.
- Morang, P.; Dutta, B.K.; Kumar, B.S.D.; Kashyap, M.P. Growth promotion and bi-control approaches of brown root rot disease of tea by Pseudomonas aeruginosa (PM 105). J. Plant Pathol. Microbiol. 2012, 3, 1–4.
- Scherling, C.; Ulrich, K.; Ewald, D.; Weckwerth, W. A metabolic signature of the beneficial interaction of the Endophyte paenibacillus sp. Isolate and in vitro-grown poplar plants revealed by metabolomics. Mol. Plant-Microbe Interact. 2009, 22, 1032–1037.
- Perva-Uzunalić, A.; Škerget, M.; Knez, Ž.; Weinreich, B.; Otto, F.; Grüner, S. Extraction of active ingredients from green tea (Camellia sinensis): Extraction efficiency of major catechins and caffeine. Food Chem. 2006, 96, 597–605.
- Namita, P.; Mukesh, R.; Vijay, K. Camellia sinensis (green tea): A review. Glob. J. Pharmacol. 2012, 6, 52–59.
- Yang, Y.; Tang, Q.; Liu, H.; Qiu, D. Tree omics and biotechnology in china. Plant OMICS 2011, 4, 288–294.
- Commisso, M.; Strazzer, P.; Toffali, K.; Stocchero, M.; Guzzo, F. Untargeted metabolomics: An emerging approach to determine the composition of herbal products. Comput. Struct. Biotechnol. J. 2013, 4, e201301007.
- Mahmood, T.; Akhtar, N.; Khan, B.A. The morphology, characteristics, and medicinal properties of Camellia sinensis’ tea. J. Med. Plants Res. 2010, 4, 2028–2033.
- Mbata, T.I. Preliminary studies of the antibacterial activities of processed kenyan and nigerian tea. Afr. J. Biotechnol. 2007, 6, 278–279.
- Ahmed, M.; Hussain, M.; Dhar, M.K.; Kaul, S. Isolation of microbial endophytes from some ethnomedicinal plants of Jammu and Kashmir. J. Nat. Prod. Plant Resour. 2012, 2, 215–220.
- Ryan, R.P.; Germaine, K.; Franks, A.; Ryan, D.J.; Dowling, D.N. Bacterial endophytes: Recent developments and applications. FEMS Microbiol. Lett. 2008, 278, 1–9.
- Joyce, A.R.; Palsson, B.O. The model organism as a system: Integrating ‘omics’ data sets. Nat. Rev. Mol. Cell Biol. 2006, 7, 198–210.
- Gehlenborg, N.; O’Donoghue, S.I.; Baliga, N.S.; Goesmann, A.; Hibbs, M.A.; Kitano, H.; Kohlbacher, O.; Neuweger, H.; Schneider, R.; Tenenbaum, D.; et al. Visualization of omics data for systems biology. Nat. methods 2010, 7, S56–S68.
- Wang, X.-C.; Zhao, Q.-Y.; Ma, C.-L.; Zhang, Z.-H.; Cao, H.-L.; Kong, Y.-M.; Yue, C.; Hao, X.-Y.; Chen, L.; Ma, J.-Q.; et al. Global transcriptome profiles of Camellia sinensis during cold acclimation. BMC Genom. 2013, 14, 415.
- Shi, C.-Y.; Yang, H.; Wei, C.-L.; Yu, O.; Zhang, Z.-Z.; Jiang, C.-J.; Sun, J.; Li, Y.-Y.; Chen, Q.; Xia, T.; et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genom. 2011, 12, 131.
- Tan, L.Q.; Wang, L.Y.; Wei, K.; Zhang, C.C.; Wu, L.Y.; Qi, G.N.; Cheng, H.; Zhang, Q.; Cui, Q.M.; Liang, J.B. Floral transcriptome sequencing for ssr marker development and linkage map construction in the tea plant (Camellia sinensis). PLoS ONE 2013, 8, e81611.
- Zhu, Q.W.; Luo, Y.P. Identification of mirnas and their targets in tea (Camellia sinensis). J. Zhejiang Univ. Sci. B 2013, 14, 916–923.
- Jaiprakash, M.R.; Pillai, B.; Venkatesh, P.; Subramanian, N.; Sinkar, V.P.; Sadhale, P.P. Rna isolation from high-phenolic freeze-dried tea (Camellia sinensis) leaves. Plant Mol. Biol. Rep. 2003, 21, 465–466.
- Wang, M.; Zhang, X.; Li, Q.; Chen, X.; Li, X. Comparative transcriptome analysis to elucidate the enhanced thermotolerance of tea plants (Camellia sinensis) treated with exogenous calcium. Planta 2018.
- Verheggen, K.; Martens, L.; Berven, F.S.; Barsnes, H.; Vaudel, M. Database search engines: Paradigms, challenges and solutions. Adv. Exp. Med. Biol. 2016, 919, 147–156.
- Chi, H.; Liu, C.; Yang, H.; Zeng, W.-F.; Wu, L.; Zhou, W.-J.; Wang, R.-M.; Niu, X.-N.; Ding, Y.-H.; Zhang, Y.; et al. Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat. Biotechnol. 2018, 36, 1059–1061.
- Ke, M.; Shen, L.; Luo, S.; Lin, L.; Yang, J.; Tian, R. Identification, quantification, and site localization of protein posttranslational modifications via mass spectrometry-based proteomics. In Modern Proteomics—Sample Preparation, Analysis and Practical Applications. Advances in Experimental Medicine and Biology; Mirzaei, M., Carrasco, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; Volume 919, pp. 345–381.
- Li, J.; Chen, J.; Zhang, Z.; Pan, Y. Proteome analysis of tea pollen (Camellia sinensis) under different storage conditions. J. Agric. Food Chem. 2008, 56, 7535–7544.
- Zhou, X.L.; Sun, P.N.; Bucheli, P.; Huang, T.H.; Wang, D. Ft-ir methodology for quality control of arabinogalactan protein (AGP) extracted from green tea (Camellia sinensis). J. Agric. Food Chem. 2009, 57, 5121–5128.
- Tugizimana, F.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. Ergosterol-induced sesquiterpenoid synthesis in tobacco cells. Molecules 2012, 17, 1698–1715.
- Krafova, K.; Jampilek, J.; Ostrovsky, I. Metabolomics in research of phytotherapeutics. Ceska Slov. Farm. Cas. Ceske Farm. Spol. Slov. Farm. Spol. 2012, 61, 21–25.
- Okada, T.; Afendi, F.M.; Altaf-Ul-Amin, M.; Takahashi, H.; Nakamura, K.; Kanaya, S. Metabolomics of medicinal plants: The importance of multivariate analysis of analytical chemistry data. Curr. Comput.-Aided Drug Des. 2010, 6, 179–196.
- Barchet, G. A Brief Overview of Metabolomics: What It Means, How It Is Measured, and Its Utilization. Available online: (accessed on 19 August 2017).
- Roessner, U.; Bowne, J. What is metabolomics all about? BioTechniques 2009, 46, 363–365.
- Shyur, L.F.; Yang, N.S. Metabolomics for phytomedicine research and drug development. Curr. Opin. Chem. Biol. 2008, 12, 66–71.
- Sharifi-Rad, M.; Nazaruk, J.; Polito, L.; Morais-Braga, M.; Rocha, J.; Coutinho, H.; Salehi, B.; Tabanelli, G.; Montanari, C.; Del, M.M.C. Matricaria genus as a source of antimicrobial agents: From farm to pharmacy and food applications. Microbiol. Res. 2018, 215, 76–88.
- Mishra, P.A.; Sharifi-Rad, M.; Shariati, M.; Mabkhot, Y.; Al-Showiman, S.; Rauf, A.; Salehi, B.; Župunski, M.; Sharifi-Rad, M.; Gusain, P. Bioactive compounds and health benefits of edible Rumex species-a review. Cell. Mol. Biol. (Noisy-le-Grand, France) 2018, 64, 27–34.
- Sharifi-Rad, M.; Roberts, T.; Matthews, K.; Bezerra, C.; Morais-Braga, M.; Coutinho, H.; Sharopov, F.; Salehi, B.; Yousaf, Z.; Del, M.M.C. Ethnobotany of the genus Taraxacum-phytochemicals and antimicrobial activity. Phytother. Res. 2018, 32, 2131–2145.
- Mishra, A.P.; Saklani, S.; Salehi, B.; Parcha, V.; Sharifi-Rad, M.; Milella, L.; Iriti, M.; Sharifi-Rad, J.; Srivastava, M. Satyrium nepalense, a high altitude medicinal orchid of indian himalayan region: Chemical profile and biological activities of tuber extracts. Cell. Mol. Biol. (Noisy-le-Grand, France) 2018, 64, 35–43.
- Azevedo, R.S.A.; Teixeira, B.S.; Sauthier, M.; Santana, M.V.A.; Dos Santos, W.N.L.; Santana, D.A. Multivariate analysis of the composition of bioactive in tea of the species Camellia sinensis. Food Chem. 2019, 273, 39–44.
- Harbowy, M.E.; Balentine, D.A.; Davies, A.P.; Cai, Y. Tea chemistry. Crit. Rev. Plant Sci. 1997, 16, 415–480.
- Punyasiri, P.A.; Abeysinghe, I.S.; Kumar, V.; Treutter, D.; Duy, D.; Gosch, C.; Martens, S.; Forkmann, G.; Fischer, T.C. Flavonoid biosynthesis in the tea plant Camellia sinensis: properties of enzymes of the prominent epicatechin and catechin pathways. Arch Biochem Biophys. 2004, 431, 22–30.
- Le Gall, G.; Colquhoun, I.J.; Defernez, M. Metabolite profiling using 1h nmr spectroscopy for quality assessment of green tea, Camellia sinensis (L.). J. Agric. Food Chem. 2004, 52, 692–700.
- Liu, L.; Li, Y.; She, G.; Zhang, X.; Jordan, B.; Chen, Q.; Zhao, J.; Wan, X. Metabolite profiling and transcriptomic analyses reveal an essential role of uvr8-mediated signal transduction pathway in regulating flavonoid biosynthesis in tea plants (Camellia sinensis) in response to shading. BMC Plant Biol. 2018, 18, 233.
- Ku, K.M.; Choi, J.N.; Kim, J.; Kim, J.K.; Yoo, L.G.; Lee, S.J.; Hong, Y.S.; Lee, C.H. Metabolomics analysis reveals the compositional differences of shade grown tea (Camellia sinensis L.). J. Agric. Food Chem. 2010, 58, 418–426.
- Lee, J.-E.; Lee, B.-J.; Chung, J.-O.; Hwang, J.-A.; Lee, S.-J.; Lee, C.-H.; Hong, Y.-S. Geographical and climatic dependencies of green tea (Camellia sinensis) metabolites: A 1h NMR-based metabolomics study. J. Agric. Food Chem. 2010, 58, 10582–10589.
- Lee, J.E.; Lee, B.J.; Hwang, J.A.; Ko, K.S.; Chung, J.O.; Kim, E.H.; Lee, S.J.; Hong, Y.S. Metabolic dependence of green tea on plucking positions revisited: A metabolomic study. J. Agric. Food Chem. 2011, 59, 10579–10585.
- Yao, L.; Caffin, N.; D’Arcy, B.; Jiang, Y.; Shi, J.; Singanusong, R.; Liu, X.; Datta, N.; Kakuda, Y.; Xu, Y. Seasonal variations of phenolic compounds in australia-grown tea (Camellia sinensis). J. Agric. Food Chem. 2005, 53, 6477–6483.
- Pongsuwan, W.; Fukusaki, E.; Bamba, T.; Yonetani, T.; Yamahara, T.; Kobayashi, A. Prediction of japanese green tea ranking by gas chromatography/mass spectrometry-based hydrophilic metabolite fingerprinting. J. Agric. Food Chem. 2007, 55, 231–236.
- Singh, H.P.; Ravindranath, S.D.; Singh, C. Analysis of tea shoot catechins: Spectrophotometric quantitation and selective visualization on two-dimensional paper chromatograms using diazotized sulfanilamide. J. Agric. Food Chem. 1999, 47, 1041–1045.
- Bokuchava, M.A.; Skobeleva, N.I. The chemistry and biochemistry of tea and tea manufacture. In Advances in Food Research; Chichester, C.O., Mrak, E.M., Stewart, G.F., Eds.; Academic Press: Waltham, MA, USA, 1969; Volume 17, pp. 215–292.
- Chakraborty, U.; Chakraborty, B.; Basnet, M. Plant growth promotion and induction of resistance in Camellia sinensis by Bacillus megaterium. J. Basic Microbiol. 2006, 46, 186–195.
- Chakraborty, U.; Chakraborty, B.N.; Chakraborty, A.P. Evaluation of Bacillus megaterium and Serratia marcescens and their bioformulations for promoting growth of Camellia sinensis. Int. J. Tea Sci. (IJTS) 2011, 8, 69–80.
- Chakraborty, U.; Chakraborty, B.N.; Chakraborty, A.P. Induction of plant growth promotion in Camellia sinensis by Bacillus megaterium and its bioformulations. World J. Agric. Sci. 2012, 8, 104–112.
- Nepolean, P.; Jayanthi, R.; Pallavi, R.V.; Balamurugan, A.; Kuberan, T.; Beulah, T.; Premkumar, R. Role of biofertilizers in increasing tea productivity. Asian Pacif. J. Trop. Biomed. 2012, 2, S1443–S1445.
- Tennakoon, P.L.K. Studies on Plant Growth Promoting Rhizomicroorganisms of Tea (Camellia sinensis (L.) Kuntze) Plants. Master’s Thesis, University of Agriculture Sciences, Dharwad, Karnataka, India, 2007.
- Erturk, Y.; Ercisli, S.; Sekban, R.; Haznedar, A.; Donmez, M.F. The effect of pgpr on rooting and growth of tea (Camellia sinensis Var. Sinensis) cuttings. Roman. Biotechnol. Lett. 2008, 13, 3747–3756.
- Nath, R.; Sharma, G.D.; Barooah, M. Screening of endophytic bacterial isolates of tea (Camellia sinensis L.) roots for their multiple plant growth promoting activities. Int. J. Agric. Environ. Biotechnol. 2013, 6, 211–215.
- Guo, B.; Wang, Y.; Sun, X.; Tang, K. Bioactive natural products from endophytes: A review. Prikl. Biokhimiia Mikrobiol. 2008, 44, 153–158.
- Tan, R.X.; Zou, W.X. Endophytes: A rich source of functional metabolites. Nat. Prod. Rep. 2001, 18, 448–459.
- Rosenblueth, M.; Martinez-Romero, E. Bacterial endophytes and their interactions with hosts. Mol. Plant-Microbe Interact. MPMI 2006, 19, 827–837.
- Shan, W.; Zhou, Y.; Liu, H.; Yu, X. Endophytic actinomycetes from tea plants (Camellia sinensis): Isolation, abundance, antimicrobial, and plant-growth-promoting activities. BioMed Res. Int. 2018, 2018, 1470305.
- Chakraborty, U.; Chakraborty, B.N.; Chakraborty, A.P. Plant growth promoting activity of Bacillus pumilus in tea (Camellia sinensis) and its biocontrol potential against poria hypobrunnea. Indian Phytopath 2013, 66, 387–396.
- Chakraborty, U.; Chakraborty, B.N.; Chakraborty, A.P.; Sunar, K.; Dey, P.L. Plant growth promoting rhizobacteria mediated improvement of health status of tea plants. Indian J. Biotechnol. 2013, 12, 20–31.
- Phukan, I.; Madhab, M.; Bordoloi, M.; Sarmah, S.R.; Dutta, P.; Begum, R.; Tanti, A.; Bora, S.; Nair, S.C.; Rai, S.; et al. Exploitation of PGP microbes of tea for improvement of plant growth and pest suppression: Anovel approach. Two Bud 2012, 59, 69–74.
- Mbata, T.I.; Debiao, L.U.; Saikia, A. Antibacterial activity of the crude extract of chinese green tea (Camellia sinensis) on listeria monocytogenes. Afr. J. Biotechnol. 2008, 7, 1571–1573.
- Hara-Kudo, Y.; Yamasaki, A.; Sasaki, M.; Okubo, T.; Minai, Y.; Haga, M.; Kondo, K.; Sugita-Konishi, Y. Antibacterial action on pathogenic bacterial spore by green tea catechins. J. Sci. Food Agric. 2005, 85, 2354–2361.
- Dong, F.; Yang, Z.; Baldermann, S.; Sato, Y.; Asai, T.; Watanabe, N. Herbivore-induced volatiles from tea (Camellia sinensis) plants and their involvement in intraplant communication and changes in endogenous nonvolatile metabolites. J. Agric. Food Chem. 2011, 59, 13131–13135.
- Zambare, V.; Bhoyte, S. Antimicrobial activity of tea (Camellia sinensis). Biomed. Pharmacol. J. 2009, 2, 173–175.
- Kumar, A.; Kumar, A.; Thakur, P.; Patil, S.; Payal, C.; Kumar, A.; Sharma, P. Antibacterial activity of green tea (Camellia sinensis) extracts against various bacteria isolated from environmental sources. Recent Res. Sci. Technol. 2012, 4, 19–23.
- Mandal, S.; DebMandal, M.; Pal, N.K.; Saha, K. Inhibitory and killing activities of black tea (Camellia sinensis) extract against salmonella enterica serovar typhi and Vibrio cholerae O1 biotype el tor serotype ogawa isolates. Jundishapur J. Microbiol. 2011, 4, 115–121.
- Rozoy, E.; Bazinet, L.; Araya-Farias, M.; Guernec, A.; Saucier, L. Inhibitory effects of commercial and enriched green tea extracts on the growth of Brochothrix thermosphacta, Pseudomonas putida and Escherichia coli. J. Food Res. 2013, 2, 1–7.
- Archana, S.; Abraham, J. Comparative analysis of antimicrobial activity of leaf extracts from fresh green tea, commercial green tea and black tea on pathogens. J. Appl. Pharm. Sci. 2011, 1, 149–152.
- Chan, E.W.; Soh, E.Y.; Tie, P.P.; Law, Y.P. Antioxidant and antibacterial properties of green, black, and herbal teas of Camellia sinensis. Pharmacogn. Res. 2011, 3, 266–272.
- Reygaert, W.C. Green tea catechins: Their use in treating and preventing infectious diseases. BioMed Res. Int. 2018, 2018, 9105261.
- Lee, J.H.; Shim, J.S.; Lee, J.S.; Kim, J.K.; Yang, I.S.; Chung, M.S.; Kim, K.H. Inhibition of pathogenic bacterial adhesion by acidic polysaccharide from green tea (Camellia sinensis). J. Agric. Food Chem. 2006, 54, 8717–8723.
- Axelrod, M.L.; Berkowitz, S.T.; Dhir, R.; Gould, V.F.; Gupta, A.; Li, E.I.; Park, J.; Shah, A.N.; Shi, K.; Tan, C.X.; et al. The Inhibitory Effects of Green Tea (Camellia Sinensis) on the Growth and Proliferation of Oral Bacteria. J. New Jersey Gov. Sch. 2010, 3, 1–19.
- Akroum, S. Antifungal activity of camellia sinensis crude extracts against four species of candida and microsporum persicolor. J. Mycol. Med. 2018, 28, 424–427.
- Simonetti, G.; Simonetti, N.; Villa, A. Increased microbicidal activity of green tea (Camellia sinensis) in combination with butylated hydroxyanisole. J. Chemother. (Florence, Italy) 2004, 16, 122–127.
- Olosunde, O.F.; Abu-Saeed, K.; Abu-Saeed, M.B. Phytochemical screening and antimicrobial properties of a common brand of black tea (Camellia sinensis) marketed in nigerian environment. Adv. Pharm. Bull. 2012, 2, 259–263.
- Diker, K.S.; Akan, M.; Hascelik, G.; Yurdakök, M. The bactericidal activity of tea against Campylobacter jejuni and Campylobacter coli. Lett. Appl. Microbiol. 1991, 12, 34–35.


