 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cynthia Mathew | + 1355 word(s) | 1355 | 2021-04-14 08:12:29 | | | |
| 2 | Karina Chen | Meta information modification | 1355 | 2021-06-15 10:22:40 | | | | |
| 3 | Karina Chen | Meta information modification | 1355 | 2021-06-15 10:23:28 | | | | |
| 4 | Karina Chen | -1 word(s) | 1354 | 2021-06-15 10:50:02 | | |
Video Upload Options
Rhinoviruses (RVs) are non-enveloped positive sense RNA viruses with a lifecycle fully contained within the cytoplasm. Despite decades of study, the details of how RVs exit the infected cell are still unclear. Current literature strongly suggests a role for necroptosis and/or autophagy in RV release, with the caveat that all the literature is based on RV-A and RV-B strains, with no studies to date examining the interaction of RV-C strains with cell death pathways.
1. Introduction
Programmed cell death is a key component of the host antiviral response, but picornaviruses, including rhinoviruses (RVs), are able to modulate cell death at different stages of virus lifecycle; inhibition of apoptosis early in infection facilitates virus survival, while induction of apoptosis later in infection may aid virus release. In addition to apoptosis, we now know that there are several kinds of programmed cell death, e.g., necroptosis, pyroptosis, ferroptosis and parthanatos; all of which are implicated in one or more virus infections [1][2][3][4]. With our increasing understanding of the complexities of cell death pathways, and the host–virus interactions during RV infection, it is becoming clear that RV components interact with, interrupt or modulate several cell-signaling cascades in the infected cell to support the virus lifecycle [1][2][3][4][5].
2. Virion Structure and Lifecycle
RVs are non-enveloped single-stranded(+) RNA viruses. The structural proteins VP1, VP2, VP3, and VP4 form an icosahedral capsid that encases the RNA genome [6]. The capsids are composed of 60 copies each of VP1-4. The VP1-3 confer strain-specific properties of immunogenicity, receptor binding and drug susceptibility to each RV isolate [7][8][6]. RV infection typically involves the following sequence (Figure 1).
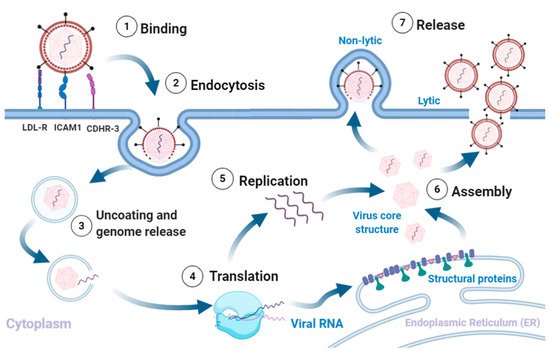
Figure 1. Rhinovirus lifecycle. (1) Binding of the virus to receptors on the plasma membrane initiates (2) endocytosis. The intake of the virion is followed by (3) uncoating of the capsid and release of the (+)ss-RNA into the host cytoplasm, which is (4) translated and processed into structural and non-structural proteins. The viral RNA dependent RNA polymerase converts the viral genome into (−)ss-RNA, that is (5) replicated into new (+)ss-RNA genomes. The new genomes become the template for translation into new viral proteins and replication into new genomes. The final stages involve (6) assembly of the structural proteins and RNA genome into capsids and (7) release of new infectious virions via lysis or non-lytic mechanisms. Adapted from [5][7][9][10]. Created with BioRender.com, accessed on 12 December 2020.
3. Rhinovirus can Modulate Apoptosis
Infection with RV-B14 in HeLa and 16HBE14o− bronchial epithelium cells exhibited typical apoptotic morphological alterations similar to those induced by puromycin [11]. The RV 3Cpro targets several cellular proteins, probably to support virus replication and limit the innate antiviral response, including apoptosis. Nuclear transport of caspases is key to the nuclear consequences of apoptosis and it is possible that the disruption of nuclear transport in RV infected cells [12][13] may serve to inhibit the progression of apoptosis; however, this remains to be investigated [14]. RV 3Cpro cleaves RIP1, a central player in pro-inflammatory, pro-survival and cell death pathways. However, subsequent cleavage of RIP1 by 3Cpro halts the progression of apoptosis. Intriguingly, markers of a necrotic cell death pathway were observed late in infection, along with membrane rupture [15]. Sudden release of calcium ions from their storage location in the Endoplasmic Reticulum (ER) can induce intrinsic apoptosis. The RV 2B protein inserts into the intracellular membranes and disrupts calcium stores, probably via leakage of calcium ions into the cytoplasm, reducing the calcium ions available to translocate to the mitochondria, resulting in apoptosis inhibition [16][17][18].
4. Autophagy and Rhinovirus Infection
The interactions between RV and autophagy are relatively understudied compared to other cell death pathways. However, it has gained attention in recent years. RV infection has been shown to induce the formation of autophagosome-like structures [19]; however, not all RV strains use autophagy in the same way or at all. A study has suggested that RV-A2, a minor group RV, induces and uses autophagy for more efficient replication, although it is still unclear as to what autophagy machinery is involved [19]. RV-A2 infected HeLa-cells showed autophagosome formation indicated both by punctate green fluorescent protein (GFP)-LC3 signal colocalized with LAMP1 staining, and an increase in LC3-II, during the later stages of infection (4 and 6 h post infection). These two points of data confirm that RV-2 infection stimulates autophagic induction [19]. However, further studies are needed to validate for different RV strains. LC3 II/LC3 I protein, an indicator of autophagosome formation, was increased during RV-A16 infection in a cell line over expressing IRAK-M; which may facilitate RV-A16 replication in cells [20]. RV-B14 infection may also induce autophagy as shown by co-localization of LAMP1 with punctate GFP-LC3 in MCF-7 cells [19]. In contrast, RV-A1a does not induce or use autophagic signaling or autophagosomes [19]. In the same study in which RV-A2 was found to induce LC3 modification, simulating autophagy, RV-A1a did not induce LC3 modification or simulate autophagy [19]. No other RV strains have been investigated for their exploitation and/or modulation of autophagy.
5. Modulation of Necrosis by Rhinovirus
Lötzerich et al., 2018 have shown 3Cpro is able to suppress apoptosis and trigger necrosis [15]. Montgomery et al., 2020 have shown a direct correlation between RV infection, increased expression of IL-1 and increased necrotic events [21]. RV was shown to induce necrosis in airway epithelial cells collected from children with and without cystic fibrosis accompanied by higher levels of inflammatory mediators IL-1α, IL-1β and IL-8 [21], suggesting necrosis as a mechanism by which RVs induce mucin accumulation and inflammation in early lung disease. Whether necrosis has a role in virus assembly or release is unclear.
6. Potential Exploitation of Necroptosis by Rhinovirus
Recent studies [22][23] have identified that RV infection inhibits caspase-dependent cell death pathways like apoptosis through the cleavage of RIP1 by 3Cpro, coinciding with impaired recruitment of active caspase 8 [23]. Inhibition of caspase 8 activity would be expected to commit the cell to necroptosis. RV infection triggers the production of several cytokines and chemokines (including IL-1, IL-6, IL-8, GM-CSF, eotaxins, and regulated upon activation normal T-cell expressed and secreted (RANTES), TNF-α, IFN-γ, and macrophage inflammatory protein (MIP)–1a [24][25][26]). Among these, TNF-α and IL-1 are known to induce the necroptotic pathway [27]. This surge in expression of cytokines and chemokines leads us to speculate that RV-induced inflammation may trigger necroptosis within the cell. However, further studies are required to address this link in more detail.
7. Parthanatos, Stress and Rhinovirus
Parthanatos is regulated by poly(ADP-ribose) polymerase 1 (PARP1), an enzyme that catalyzes DNA base excision repair [28]. RV-1B infection of human primary bronchial epithelial cells (pBECs) exposed to cigarette smoke or other oxidative stressors reduced mitochondrial respiration, increased proton leak and subsequently increased pro-inflammatory cytokines [29]. The oxidative stress caused by the external stimuli, including RV infection, lead to increased expression of cytochrome C [29]. This suggests parthanatos could be one of the mechanisms by which RV infections, aggravated by external stimuli such as cigarette smoke, can induce exacerbations in asthma and chronic obstructive pulmonary disease (COPD) [29].
8. Conclusion
That RVs manipulate cell death pathways to favour viral replication and propagation, is clear [30]. This manipulation may correlate with the severity of disease and exacerbations of pre-existing lung conditions [31]. Accumulating literature shows that RVs inhibit apoptosis early in infection via cleavage of RIP1 and by disrupting nucleocytoplasmic trafficking; although the latter has not been directly linked with apoptosis inhibition. Recent studies implicate necroptosis as a potential mechanism by which RVs exit the host cell, accompanied by release of proinflammatory chemokines and cytokines. RIP1, which is cleaved in RV infection, is a master regulator, positioned to commit the cell to prosurvival, apoptotic or necroptotic pathway. RV infection induces caspase 8 dependent apoptotic pathway, which is quickly inhibited by cleavage of RIP1, possibly committing the cell to necroptosis. This supposition is supported by the observation of necrotic markers in the absence of late-stage necrosis in RV infected cells. RVs appear to modulate autophagy in a strain, maybe species, specific manner. Parthanatos is one of the mechanisms by which RV infections may induce exacerbations in asthma and COPD. Intriguingly, cell death related targets of 3Cpro identified so far (e.g., p62/SQSTM1) are involved in multiple pathways, possibly suggesting a fine modulation of several pathways by RVs that may vary through the lifecycle to enable optimal virus replication, assembly and release.
References
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.
- Kuo, C.-Y.; Chiu, V.; Hsieh, P.-C.; Huang, C.-Y.; Huang, S.J.; Tzeng, I.S.; Tsai, F.-M.; Chen, M.-L.; Liu, C.-T.; Chen, Y.-R. Chrysophanol attenuates hepatitis B virus X protein-induced hepatic stellate cell fibrosis by regulating endoplasmic reticulum stress and ferroptosis. J. Pharmacol. Sci. 2020, 144, 172–182.
- Suwanmanee, S.; Luplertlop, N. Immunopathogenesis of Dengue Virus-Induced Redundant Cell Death: Apoptosis and Pyroptosis. Viral Immunol. 2017, 30, 13–19.
- Kim, J.H.; Kim, J.; Roh, J.; Park, C.S.; Seoh, J.Y.; Hwang, E.S. Reactive oxygen species-induced parthanatos of immunocytes by human cytomegalovirus-associated substance. Microbiol. Immunol. 2018, 62, 229–242.
- Croft, S.N.; Walker, E.J.; Ghildyal, R. Picornaviruses and Apoptosis: Subversion of Cell Death. mBio 2017, 8, e01009-17.
- Baggen, J.; Thibaut, H.J.; Strating, J.; van Kuppeveld, F.J.M. The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Microbiol. 2018, 16, 368–381.
- Jacobs, S.E.; Lamson, D.M.; St George, K.; Walsh, T.J. Human rhinoviruses. Clin. Microbiol. Rev. 2013, 26, 135–162.
- Stobart, C.C.; Nosek, J.M.; Moore, M.L. Rhinovirus Biology, Antigenic Diversity, and Advancements in the Design of a Human Rhinovirus Vaccine. Front. Microbiol. 2017, 8.
- Tuthill, T.J.; Groppelli, E.; Hogle, J.M.; Rowlands, D.J. Picornaviruses. Curr. Top. Microbiol. Immunol. 2010, 343, 43–89.
- Jiang, P.; Liu, Y.; Ma, H.-C.; Paul, A.V.; Wimmer, E. Picornavirus morphogenesis. Microbiol. Mol. Biol. Rev. 2014, 78, 418–437.
- Deszcz, L.; Gaudernak, E.; Kuechler, E.; Seipelt, J. Apoptotic events induced by human rhinovirus infection. J. Gen. Virol. 2005, 86, 1379–1389.
- Ghildyal, R.; Jordan, B.; Li, D.; Dagher, H.; Bardin, P.G.; Gern, J.E.; Jans, D.A. Rhinovirus 3C Protease Can Localize in the Nucleus and Alter Active and Passive Nucleocytoplasmic Transport. J. Virol. 2009, 83, 7349.
- Gustin, K.E.; Sarnow, P. Inhibition of nuclear import and alteration of nuclear pore complex composition by rhinovirus. J. Virol. 2002, 76, 8787–8796.
- Ferrando-May, E. Nucleocytoplasmic transport in apoptosis. Cell Death Differ. 2005, 12, 1263–1276.
- Lotzerich, M.; Roulin, P.S.; Boucke, K.; Witte, R.; Georgiev, O.; Greber, U.F. Rhinovirus 3C protease suppresses apoptosis and triggers caspase-independent cell death. Cell Death Dis. 2018, 9, 272.
- Brisac, C.; Téoulé, F.; Autret, A.; Pelletier, I.; Colbère-Garapin, F.; Brenner, C.; Lemaire, C.; Blondel, B. Calcium Flux between the Endoplasmic Reticulum and Mitochondrion Contributes to Poliovirus-Induced Apoptosis. J. Virol. 2010, 84, 12226–12235.
- Campanella, M.; de Jong, A.S.; Lanke, K.W.; Melchers, W.J.; Willems, P.H.; Pinton, P.; Rizzuto, R.; van Kuppeveld, F.J. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem. 2004, 279, 18440–18450.
- de Jong, A.S.; de Mattia, F.; Van Dommelen, M.M.; Lanke, K.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. Functional analysis of picornavirus 2B proteins: Effects on calcium homeostasis and intracellular protein trafficking. J. Virol. 2008, 82, 3782–3790.
- Klein, K.A.; Jackson, W.T. Human rhinovirus 2 induces the autophagic pathway and replicates more efficiently in autophagic cells. J. Virol. 2011, 85, 9651–9654.
- Wu, Q.; van Dyk, L.F.; Jiang, D.; Dakhama, A.; Li, L.; White, S.R.; Gross, A.; Chu, H.W. Interleukin-1 receptor-associated kinase M (IRAK-M) promotes human rhinovirus infection in lung epithelial cells via the autophagic pathway. Virology 2013, 446, 199–206.
- Montgomery, S.T.; Frey, D.L.; Mall, M.A.; Stick, S.M.; Kicic, A.; Arest, C.F. Rhinovirus Infection Is Associated With Airway Epithelial Cell Necrosis and Inflammation via Interleukin-1 in Young Children With Cystic Fibrosis. Front. Immunol. 2020, 11.
- Deszcz, L.; Gaudernak, E.; Kuechler, E.; Seipelt, J. Apoptotic events induced by human rhinovirus infection. J. Gen. Virol. 2005, 86, 1379–1389.
- Croft, S.N.; Walker, E.J.; Ghildyal, R. Human Rhinovirus 3C protease cleaves RIPK1, concurrent with caspase 8 activation. Sci. Rep. 2018, 8, 1569.
- Kelly, J.T.; Busse, W.W. Host immune responses to rhinovirus: Mechanisms in asthma. J. Allergy Clin. Immunol. 2008, 122, 671–682.
- Gern, J.E.; Dick, E.C.; Lee, W.M.; Murray, S.; Meyer, K.; Handzel, Z.T.; Busse, W.W. Rhinovirus enters but does not replicate inside monocytes and airway macrophages. J. Immunol. 1996, 156, 621–627.
- Jackson, D.J.; Makrinioti, H.; Rana, B.M.; Shamji, B.W.; Trujillo-Torralbo, M.B.; Footitt, J.; Jerico, D.-R.; Telcian, A.G.; Nikonova, A.; Zhu, J.; et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am. J. Respir. Crit. Care Med. 2014, 190, 1373–1382.
- Calvén, J.; Akbarshahi, H.; Menzel, M.; Ayata, C.K.; Idzko, M.; Bjermer, L.; Uller, L. Rhinoviral stimuli, epithelial factors and ATP signalling contribute to bronchial smooth muscle production of IL-33. J. Transl. Med. 2015, 13, 281.
- Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Mitochondrial and nuclear cross talk in cell death: Parthanatos. In Annals of the New York Academy of Sciences; Wiley-Blackwell: Hoboken, NJ, USA, 2008; Volume 1147, pp. 233–241.
- Wark, P.; Pathinyake, P.; Hsu, A.; Parsons, K.; Wood, L. Effect of oxidative stress and rhinovirus infection on mitochondrial/endoplasmic reticular function in human primary bronchial epithelial cells. Eur. Respir. J. 2016, 48, PA3989.
- Danthi, P. Viruses and the Diversity of Cell Death. Annu. Rev. Virol. 2016, 3, 533–553.
- Papi, A.; Contoli, M. Rhinovirus vaccination: The case against. Eur. Respir. J. 2011, 37, 5.


