Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Thorsten Stiewe | + 4473 word(s) | 4473 | 2021-06-04 04:40:08 | | | |
| 2 | Vivi Li | Meta information modification | 4473 | 2021-06-10 10:34:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Stiewe, T. The Mutant p53 Secretome. Encyclopedia. Available online: https://encyclopedia.pub/entry/10717 (accessed on 07 February 2026).
Stiewe T. The Mutant p53 Secretome. Encyclopedia. Available at: https://encyclopedia.pub/entry/10717. Accessed February 07, 2026.
Stiewe, Thorsten. "The Mutant p53 Secretome" Encyclopedia, https://encyclopedia.pub/entry/10717 (accessed February 07, 2026).
Stiewe, T. (2021, June 10). The Mutant p53 Secretome. In Encyclopedia. https://encyclopedia.pub/entry/10717
Stiewe, Thorsten. "The Mutant p53 Secretome." Encyclopedia. Web. 10 June, 2021.
Copy Citation
p53 suppresses tumorigenesis by activating a plethora of effector pathways. While most of these operate primarily inside of cells to limit proliferation and survival of incipient cancer cells, many extend to the extracellular space. In particular, p53 controls expression and secretion of numerous extracellular factors that are either soluble or contained within extracellular vesicles such as exosomes. As part of the cellular secretome, they execute key roles in cell-cell communication and extracellular matrix remodeling. Mutations in the p53-encoding TP53 gene are the most frequent genetic alterations in cancer cells, and therefore, have profound impact on the composition of the tumor cell secretome.
p53
tumor suppressor
secretome
extracellular vesicles
exosomes
tumor microenvironment
pre-metastatic niches
metastasis
1. Introduction
Originally discovered at the highpoint of tumor virus research and initially classified as an oncogene, the tumor suppressor p53 is now considered one of the most critical protectors of the human genome and a central component of a multiplex molecular network of signaling cascades [1][2][3][4]. Subsequent to various cellular insults that include DNA damage, oxidative stress, and oncogenic signaling, p53 is activated and functions as a sequence-specific transcription factor, setting in motion pathways for DNA repair, cell cycle arrest, apoptosis, and senescence [4][5][6][7]. Interestingly, p53-like genes resembling mammalian p53 in sequence and function were first identified in the evolution of modern-day simple invertebrate organisms that are not inclined to tumor development [4][8][9]. Similar to mammalian p53, these genes trigger cell death upon stress stimuli; however, they are only expressed in the germline, suggesting that p53 did not evolve to prevent cancer but rather to protect cells from genomic instability and replicative stress [4][10][11]. p53’s ability to defeat somatic tumors at advanced age evolved later synchronously with the evolution of multicellular life [10][11][12].
p53’s potency in suppressing inappropriate clonal outgrowth is unparalleled and clearly evident from the severe cancer susceptibility of mice and men with engineered or inherited mutations in p53, respectively [13][14][15]. To overcome p53-mediated tumor suppression, cancer cells have developed multiple tactics to disarm p53 and, thereby, promote their own survival and expansion. The most direct way is certainly through TP53 gene mutations, and cancer genome sequencing projects have provided undeniable evidence showing that TP53 alterations are the most frequent events in human cancers [16][17][18]. TP53 is now known to be hit largely by missense mutations, although deletions, truncations, and frameshift mutations have also been reported [16][18]. Among the missense mutations, roughly 80% affect residues within the p53 DNA-binding core domain, where several mutational hotspots have been acknowledged [16][18]. These missense mutants have lost their ability to bind to the established p53-responsive DNA elements and launch the respective tumor suppressive programs (loss of function, LOF). In addition, missense mutants bind and inactivate wild-type proteins expressed from a nonmutated allele (dominant-negative effect, DN), and many acquire new neomorphic activities (gain of function, GOF) that boost cancer cell growth, survival, expansion, and spread in many different ways [19][20][21][22][23]. For instance, mutant p53 has been shown to control several tumor cell-autonomous processes beneficial for tumor cell survival under adverse conditions, including regulation of energy metabolism, response to proapoptotic signals, and adaptation to oxidative stress [21][24].
2. TP53 Mutations
The progress with massively parallel sequencing of tumor genomes in the past decade has provided an unprecedented insight into the numerous ways in which the TP53 locus is altered in tumors and how this unique TP53 “mutome” translates into functional consequences, leading ultimately to more aggressive tumorigenesis and a poor patient outcome [18][25].
2.1. Classes of TP53 Mutations
TP53 mutations are dispersed throughout all exons with a striking preference for the central region encoding the DNA-binding core domain. The most common (72.7%) and well-characterized TP53 mutations among the 80,400 cancer cases reported in the Universal Mutation Database (UMD) are missense mutations in the DNA-binding domain (DBD), signifying that DNA binding is crucial for the tumor suppressive function [16][26]. Six hotspot residues within the DBD (R175, G245, R248, R249, R273, and R282) are hit most frequently. Depending on whether the corresponding residues are involved in DNA contact or structure maintenance, mutant proteins are categorized as contact (R273H, R248Q, and R248W) or conformational (R175H, G245S, R249S, and R282H) [27][28]. Contact mutants derive from missense mutations in residues responsible for direct contact with the DNA sequences forming p53 response elements in target gene promoters and have an intact native fold [29][30][31]. Conformational mutations result in the disruption of the p53 protein structure by decreasing the already low folding stability of the DBD, leading to its denaturation and often aggregation at body temperature [27]. Nevertheless, the distinction between these two mutation categories is somewhat arbitrary, as there are p53 mutants that, in principle, fit in both (e.g., R248Q) [27][32]. In addition, there are DBD mutations that do not fit into this bipartite classification, such as cooperativity mutations which influence the formation of the DNA-bound tetramer without any alterations at the DNA-binding surface or the overall folding and stability of the DBD [33][34][35][36][37][38][39][40][41][42][43].
2.2. Tumor Promotion by TP53 Mutations
The notable preference for p53 missense mutations in human cancers led to the appreciation that mutant p53 proteins convey a selective advantage during tumorigenesis. On the one hand, mutant p53 can disable wild-type p53 expressed from the second allele through oligomerization (dominant-negative effect, DN) [44][45]. On the other, p53 mutants can acquire novel oncogenic functions [46]. Supporting the idea of a pro-tumorigenic gain of oncogenic activity, overexpression of mutant p53 in murine or human cells with no endogenous p53 was reported to promote malignant transformation [47][48]. Remarkably, mice carrying the R172H allele alone or in combination with wild-type p53 exhibited similar survival compared to mice homo- or heterozygous for a p53-null allele [49][50]. They did, however, present a more diverse spectrum of more aggressive and metastatic tumors, including a higher incidence of carcinomas [49][50]. In parallel, humanized p53 knock-in mice (HUPKI) where the DNA-binding domain of the p53 locus was partly substituted by the corresponding human sequence with or without hotspot mutations resulted in similar conclusions [51][52]. Notably, a recent study also reported that mouse mutant equivalents of R175H and R248W lead to rapid breast cancer development driving a parallel evolutionary pattern of metastases [53]. Consistent with these animal studies, Li-Fraumeni patients carrying TP53 missense mutations in the germline develop cancer significantly earlier than patients with nonsense or frame-shift mutations that result in loss of p53 protein expression by nonsense-mediated mRNA decay [54][55]. Overall, these in vivo genetic studies effectively portrayed the crucial role of mutant p53 proteins in the development and metastatic progression of tumors and laid the foundations for the mutant p53 GOF concept. Of note, the GOF concept is not undisputed, and two recent large-scale functional studies of the p53 mutome have revealed the dominant-negative effect as the primary unit of selection for TP53 missense mutations [56][57].
Even though we often refer to mutated p53 proteins as “mutant p53” (or short, “mutp53”), we are becoming increasingly aware of the incredible functional diversity within the p53 mutome [21][25]. p53 cancer mutants differ dramatically regarding the loss of wild-type activity, the degree of dominant-negative activity, and the quantity and quality of neomorphic (GOF) activities [15][22][38][58]. With respect to the numerous ways that p53 mutants operate to alter the tumor cell secretome, one may therefore differentiate between mechanisms that are caused by the abrogation or inhibition of wild-type p53 functions (loss-of-function and dominant-negative activities) or true neomorphic mutant p53 activities (Figure 1).
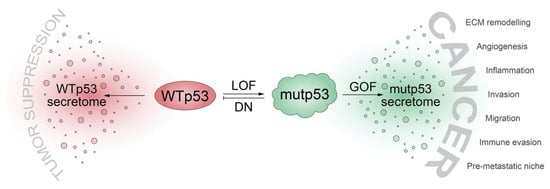
Figure 1. TP53 mutations shift the cellular secretome from a tumor suppressive secretome driven by wild-type p53 (WTp53) to a cancer-promoting secretome through: loss of WTp53 function (LOF), dominant-negative (DN) inhibition of WTp53 by mutant p53 (mutp53), and gain of neomorphic functions with oncogenic properties (GOF). ECM, extracellular matrix.
3. Mutant p53 GOF Mechanisms Altering the Tumor Secretome
Although most p53 mutations largely abrogate the DNA binding activity of p53, directly or in a dominant-negative fashion, mutant p53 proteins are far from transcriptionally inert and can modulate gene expression indirectly by binding and regulating numerous transcription factors and chromatin remodelers (Figure 4) [19][59]. Furthermore, p53 mutants can interact with cellular proteins involved in signal transduction and secretory pathways resulting in altered protein secretion.
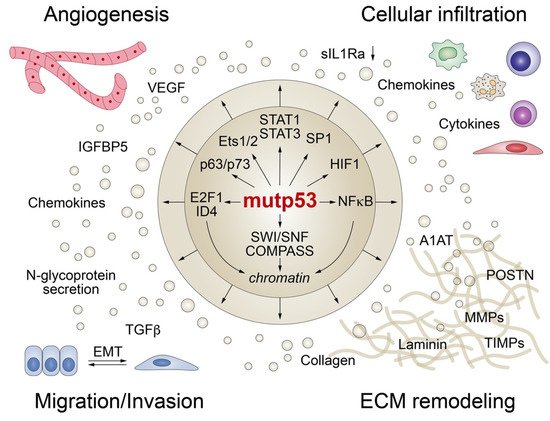
Figure 4. Mutant p53 shapes the tumor cell secretome at the transcriptional level. Mutant p53 interacts with transcription factors and/or chromatin-modifying enzymes (SWI/SNF and COMPASS) to promote angiogenesis, trigger epithelial-mesenchymal transition (EMT), remodel the extracellular matrix (ECM), and modulate the composition and function of the cellular stroma infiltrate.
3.1. Mutant p53 Modifies the Activity of Other TFs
Although there is some evidence that mutant p53—similar to wild-type p53—binds DNA in a sequence-unspecific but structure-specific manner [60][61][62][63], mutant p53 seems to interact with DNA primarily indirectly through physical association with other transcription factors that recruit mutant p53 to their respective target genes [19][22][24][64]. Although many of the TFs bound by mutant p53 also interact with wild-type p53, the functional outcome is often opposite. As these mutant p53 activities are dependent on other transcription factors, many of which are expressed in a cell- or stimulus-specific manner, there is considerable context-specificity. In addition, the interactions between mutant p53 and other TFs are structurally quite diverse, and therefore, also influenced by the type of mutation (conformational/contact) or the modified residue.
3.1.1. p53 Family Members
One of the best-studied mechanisms by which p53 mutants impact on gene expression is via co-aggregation and sequestration of the two tumor suppressive family members p63 and p73 [49][65][66][67]. While p63 and p73 have been demonstrated to form homo- and heterotetramers with each other, neither forms heterotetramers with wild-type p53 [68]. Instead, mutant p53 interacts with the family members through the DNA-binding core domain [66][69]. Specific interactions between mutant p53 and p63/p73 may result from the structural changes in the DNA-binding core domain that are characteristic for missense mutations of the conformational class, but p53 contact mutants have also been shown to interact with p63 and p73 [70][71]. Given that p63 and p73 are sequence-specific transcription factors, interaction with p53 mutants alters their activity at their target genes positively or negatively depending on promoter context [66]. Both activation and repression have been demonstrated to increase invasion and metastasis. For example, certain p53 mutants but not wild-type p53 may interact and inhibit the family member TAp63 to control pro-invasive and pro-metastatic transcriptional profiles via regulation of Sharp1, Cyclin G2, and miR-155 [66][69][72][73][74]. Mutp53-p63 interaction also drives invasion and metastasis by stimulating TGF-β signaling, recycling of EGFR and integrins via RCP, and downregulating Dicer-mediated processing of anti-metastatic miRNAs [24][69][73][75][76]. Importantly, mutant p53 can use p63 as a molecular chaperone to tether to its target gene’s promoters and alter gene expression profiles to promote oncogenesis [77]. Expression profiling of six different inducible mutant p53 cell lines revealed a core set of 59 target genes highly enriched with p63 target genes that encode secreted protein products and form a pro-invasive secretome [77].
Furthermore, quantitative proteomic characterization of the secretome from non-small cell lung cancer cells has demonstrated the power of mutant p53 to drive expression of secreted proteins that function in either autocrine or paracrine signaling to promote migration and invasion of tumor cells [78]. Among them were BIGH3, an ECM protein that modulates cell adhesion, alpha-1 antitrypsin (A1AT or SERPINA1), a secreted serine protease inhibitor, and various epithelial-mesenchymal transition markers. A1AT was upregulated in a transcriptional manner via cooperation of mutant p53 with the family member p63 and identified as a critical and indispensable target for cell migration and invasion in vitro and in vivo [78]. A1AT expression correlated with increased tumor stage and shorter survival of lung cancer patients and was predominant at the boundary between tumor and stroma, suggesting a primary function in tumor-stroma crosstalk [78]. Additionally, in several other human cancer types, A1AT expression was shown to correlate with invasion and metastasis [79][80][81], and high levels of secreted A1AT in blood and urine samples from cancer patients were associated with worse patient outcomes [79][82]. Although the pleiotropic mechanisms of A1AT in tumorigenesis are not fully understood, A1AT presumably functions at least in part via inhibiting the activity of enzymes present in the extracellular environment. For example, inhibition of proteases, like mast cell chymase and leukocyte elastase, can influence TGF-β availability in the ECM, and thereby, affect tumor cell invasion [83]. In addition, A1AT has been reported to induce the release of the angiopoietin-like protein 4 via a peroxisome proliferator-activated receptor-dependent pathway [84], indicating that protease inhibitor-independent functions likely contribute to its activity as a mediator of mutant p53’s pro-tumorigenic activity.
3.1.2. NF-κB
Other examples include complexes of mutant p53 with the TFs NF-κB, NF-Y, Ets-1 and Ets-2, SP1, VDR, E2F1, ID4, and SREBP [22]. Apart from the crosstalk of NF-κB and wild-type p53 described in the previous section, p53 mutations have been shown to modulate NF-κB-driven inflammatory gene expression in multiple ways independent of their effect on wild-type p53. First, mutant p53 was shown to enhance induction of NF-κB activity by TNF-α associated with enhanced nuclear accumulation of NF-κB [85]. Similarly, certain p53 mutants augment NF-κB activity, resulting in increased CXCL5, CXCL8, and CXCL12 chemokine expression crucial for tumor cell migration [86]. Interestingly, CXCL8 itself has the capacity to trigger NF-kB activation, suggesting that mutant p53 might initiate the formation of an autocrine or paracrine feedback loop [87]. Furthermore, wide transcriptome profiling of various cancer cell lines revealed a cancer-related gene signature (CGS) encoding mainly pro-cancerous secreted molecules that is synergistically upregulated by p53 inactivation and oncogenic HrasV12 expression [88][89]. The identified CGS comprises many ECM-related proteins, like TFPI2, MMP3, PRSS2, C1QTNF1, and ADAMTS8, but also various CXC chemokines and cytokines [89]. Interestingly, various p53 mutations were found to promote the secretion of CGS factors via distinct mechanisms and to different extents. Loss of wild-type p53 reduces expression of the p53 target gene BTG2, which normally represses Hras by reducing its GTP-loaded state [88]. The zinc region conformational mutants R175H and R179R also augment Hras activity by inhibiting BTG2, while contact mutants R248Q and R273H induce a significantly higher CGS expression by boosting NF-κB activation [90]. Notably, the L3 loop conformation mutant G245S does not seem to affect the CGS, which might be explained by the observation that this mutation partially retains some wild-type p53 characteristics [90]. In a mouse model for colorectal cancer induced by chronic inflammation, mutant p53—but not loss of p53—triggered a pronounced NF-κB-mediated inflammatory response acting as a driver of colorectal tumorigenesis and invasion [91].
Mechanistically, early chromatin immunoprecipitation (ChIP-on-Chip) studies revealed an overrepresentation of NF-κB binding sites among mutant p53-bound sequences [92]. Later, in-depth ChIP-seq coupled with transcriptome (RNA-seq and GRO-seq) analyses demonstrated a global overlap in the binding of p53 mutants and NF-κB/p65 that drives alterations in enhancer and gene activation in response to chronic TNF-α signaling [93]. p53 mutants were shown to directly interact with NF-κB/p65 impacting on each other’s DNA binding profile [93]. Simultaneous and cooperative binding of mutant p53 and NF-κB was shown to be required for RNA polymerase II recruitment, synthesis of enhancer RNAs, and activation of various tumor-promoting secreted factors, including MMP9 and CCL2 [93]. Of note, mutant p53 was also shown to directly bind and activate the promoters of the NF-κB target genes CXCL-1 (GRO1) without changes in NF-κB signaling, presenting an alternative NF-kB-independent mechanism for the induction of cytokine secretion [94].
Apart from these nuclear functions of mutant p53, cytoplasmic activities were also shown to impinge on inflammatory NF-κB signaling. In response to TNF-α, mutant p53 sustains activation of NF-κB while dampening activation of JNK by directly binding and inactivating the cytoplasmic RasGAP DAB2IP, a tumor-suppressive protein that switches the TNF-α response from NF-κB to JNK signaling [95]. As a consequence, the mutp53-DAB2IP interaction enhances the invasive behavior of cancer cells exposed to an inflammatory microenvironment and represents an indirect means of how neomorphic properties of mutant p53 enhance pro-tumorigenic NF-κB signaling [95].
3.1.3. STAT Transcription Factors
In a mouse model of chronic colitis-associated colorectal cancer, p53 mutants were found to interact with the signal-transducing STAT3 TF and enhance its transcriptional activity by displacing the STAT3-inhibitory SHP2 phosphatase [96]. This mutp53/STAT3 axis was shown to be crucial for colorectal cancer cell survival and a pro-invasive epithelial-to-mesenchymal transition (EMT) phenotype [96]. Intriguingly, mutp53-activated STAT3 signaling occurred in the absence of an NF-κB-driven secretory response observed in different colitis-associated colorectal cancer models [97][91][96]. It can be speculated that the distinct microenvironmental phenotypes reflect differences in experimental protocols (acute/transient versus chronic inflammation) and p53 mutation status (null versus mutant alleles and global versus tissue-specific mutations), further illustrating the highly context-specific effects of p53 mutations on the tumor secretome.
Recent studies have also revealed a connection of mutant p53 to STAT1 signaling in the context of matricellular protein secretion [98]. Matricellular proteins are distinct from the fibrillar ECM proteins with primary structural roles and have emerged as important biological mediators of cell function by promoting ECM remodeling and initiating downstream signaling via integrins or RTKs [99]. The matricellular protein periostin (POSTN) was shown to be important for cancer cell dissemination and metastatic colonization, and, accordingly, POSTN overexpression has been observed in numerous advanced stage cancers, including breast and NSCLC, where the p53 mutation rate is high [98]. In a study on esophageal cancer, the p53 missense mutant R175H was shown to cooperate with POSTN to enhance STAT1 signaling [98]. The STAT1-regulated gene network includes numerous secreted factors known to promote tumor progression via maintenance of a pro-inflammatory microenvironment, suggesting that mutp53-POSTN cooperation contributes to a permissive tumor microenvironment that facilitates invasion.
3.1.4. SP1
Furthermore, p53 mutants interact with the SP1 transcription factor at the EGFR promoter, resulting in promoter activation by histone acetyltransferase recruitment [100]. In addition, mutp53/SP1 complexes also mediate transactivation of the ectonucleoside triphosphate diphosphohydrolase 5 (ENTPD5) gene, which encodes an endoplasmic reticulum (ER)-resident UDPase [101][102][103][104]. By cleaving UDP to UMP in the ER, ENTPD5 promotes import of UDP-glucose into the ER, which is essential for the proper folding of nascent N-glycoproteins by the calnexin/calreticulin (CANX/CALR) chaperone cycle. Since the majority of secreted and membrane-bound proteins are N-glycoproteins, and therefore, strongly depend on processing by the CANX/CALR cycle, upregulation of the N-glycoprotein-folding capacity by mutant p53 can be expected to heavily boost protein secretion [105][106]. In addition, upregulation of CANX/CALR cycle activity modifies the composition of the secretome, as various N-glycoproteins rely on chaperones to a different extent [105][106][107]. While it was shown that mutp53-induced ENTPD5 expression is crucial for cell migration, invasion, and lung colonization by p53-mutant breast cancer cells, the relevant N-glycoproteins affected by the mutp53-ENTPD5 axis remain unknown.
3.2. Mutant p53 Modifies the Activity of Chromatin-Regulators
Another tactic by which p53 mutations control the tumor secretome at the transcriptome level is through modulation of the chromatin landscape. For example, p53 mutant tumor cells show an elevated expression of the chromatin-regulatory COMPASS (complex proteins associated with Set1) complex subunits MLL1 (KMT2A), MML2 (KMT2D), and MOZ (KAT6A), which possess histone methyl- and acetyl-transferase activities, respectively [108]. Similarly, as in the previous section, mutant p53-mediated upregulation of these enzymatic activities was shown to involve mutp53-Ets-2 interactions for recruitment of the mutated p53 protein to the MLL1, MLL2, and MOZ gene promoters. COMPASS inhibitors selectively compromise growth of p53-mutated cells, indicating a specific requirement for mutp53-dependent chromatin regulation for the maintenance of a highly malignant phenotype. Although it is known that the COMPASS complex is critical for the expression of numerous inflammatory target genes [109], the impact of the mutp53-COMPASS axis on the tumor secretome remains to be investigated.
In addition, mutant p53 regulates expression of numerous genes in a manner dependent on the SWI/SNF nucleosome remodeling complex [110][111]. As described above, the SWI/SNF complex can be recruited to genes by a mutp53/HIF1 complex assembled in hypoxic tumor cells [110]. Vice versa, the SWI/SNF complex can recruit mutant p53 to promoters, as shown for VEGFR2 [111]. This results in the remodeling of the promoter, the maintenance of an open conformation, and the transcriptional activation of the corresponding VEGFR2 gene [111]. Genome-wide expression profiling revealed that roughly 50% of the genes regulated by mutant p53 were also regulated by SWI/SNF, indicating that mutant p53 harnesses SWI/SNF to remodel a vast variety of promoters into a transcriptionally active conformation, including important secreted factors such as IGFBP5, ceruloplasmin, and mammaglobin-A [111]
3.3. Other Transcriptional Mechanisms
Mutant p53 was described to enhance interleukin-1 (IL-1) signaling by repressing the promoter of the secreted IL-1 receptor antagonist sIL-1Ra [112]. Cytokines such as interleukins, interferons, or TNF-α function as molecular messengers, allow immune cells to communicate with one another, and are involved in tumor immune-surveillance in an efficiently regulated multifaceted, pleiotropic, and redundant manner [113]. IL-1, for example, is secreted by stromal cells and infiltrating leukocytes during inflammation and is intrinsically and extrinsically involved in cancer pathology [114]. Antagonists of IL-1 bind to IL-1 receptors without conveying any stimulating signals, thus blocking the pro-inflammatory signaling by IL-1 [115]. Mutant p53 physically interacts with the sIL-1Ra promoter and, together with the transcriptional co-repressor MAFF (v-MAF musculoaponeurotic fibrosarcoma oncogene family, protein F), represses gene transcription [112]. Thereby, mutant p53 contributes to a pro-inflammatory tumor microenvironment and sustains IL-1-driven tumor malignancy [112].
Mutant p53 also controls the activity of matrix metalloproteinases (MMPs), which play a critical role in cancer cell invasion by degrading several ECM constituents [116][117]. In tumor types, where p53 mutants accumulate to high levels, expression of tissue inhibitors of MMPs such as TIMP-3 is frequently repressed [118][119][120]. Two different p53 mutants (R248W and D281G) have been shown to inhibit TIMP-3 transcription [118]. In this study, it was demonstrated that overexpression of either of the two p53 mutants in human colon carcinoma cells leads to promoter repression of TIMP-3, resulting in reduced expression and elevated MMP activity [118]. This ultimately causes increased degradation of the extracellular matrix and basal lamina associated with increased metastatic potential. Interestingly, although wild-type p53 may also repress TIMP-3, the physiologically low levels of wild-type p53 in tissues probably allows TIMP-3 to remain highly expressed [118]. Accordingly, p53 mutant proteins are stabilized and accumulate at very high levels in human tumors, reducing TIMP-3 expression and promoting ECM turnover [118]. Of note, direct binding of mutant p53 to the TIMP-3 promoter was not observed [118], suggesting an indirect mechanism of promoter regulation that remains to be investigated.
Furthermore, mutant p53 constitutively interacts with PML, unlike the transient stress-induced association of PML with wild-type p53 [121][122]. Importantly, PML facilitates mutant p53 to aberrantly activate genes, possibly in the context of the hijacked transcription factor NF-Y [121][122]. In support of a functional interplay, oncogenic functions of mutant p53 are attenuated by PML depletion, and the tumor profile of mice with p53 mutations is shifted when PML is inactivated [121][123]. The exact role of the mutp53/PML axis for the tumor cell secretome, however, remains to be delineated.
3.4. Mutant p53 Modifies Exosomal Protein Secretion
Apart from stimulating the expression of secreted proteins or proteins involved in the classical secretory pathway, mutant p53 also exerts effects on protein secretion via extracellular vesicles (EV), comprising plasma membrane-derived “ectosomes” (microparticles/microvesicles) and “exosomes” of endosomal origin (Figure 5) [124]. Exosomes are small (30–200 nm in diameter), single-membrane organelles secreted by cells and enriched in DNA, coding and noncoding RNAs, proteins, and lipids [125][126]. Exosomes are released from a variety of cells, including fibroblasts and tumors, and their activities are diverse, ranging from remodeling of the ECM to cell-cell communication. Accordingly, they are involved in numerous processes, including angiogenesis, apoptosis, antigen presentation, inflammation, and cancer [125][126]. The production of exosomes, as well as their molecular cargo, is affected by external stress signals, such as oxidative stress or ionizing radiation. Being a critical cellular stress-responsive transcription factor, it comes as no surprise that wild-type p53 also plays major roles in exosome biogenesis by regulating both secretion and cargo [127][128]. For instance, wild-type p53 enhances exosome production in response to DNA-damage via transcriptional upregulation of TSAP-6 and CHMP4C [127][128]. In addition, p53 alters the exosomal content and size in colorectal carcinoma cells via HGS, a key component of the endosomal sorting complex [129]. Intriguingly, loading of EVs with anti-metastatic cargo, such as metalloproteinase inhibitor TIMP-3, was shown to be dependent on p53 acetylation by the BAG6/CBP/p300 complex followed by recruitment of the ESCRT (endosomal sorting complex required for transport) machinery [129].
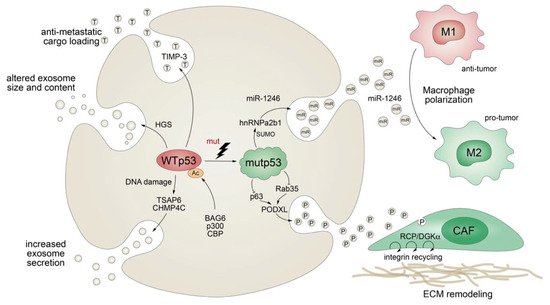
Figure 5. Mutant p53 shapes the tumor cell secretome at the exosome level. TP53 mutations abrogate wild-type p53-mediated effects on exosome secretion, size, and cargo and actively control exosomal secretion of miR-1246 and podocalyxin (PODXL), thereby affecting macrophage M1-M2 polarization and extracellular matrix (ECM) remodeling by cancer-associated fibroblasts (CAF).
While evidence for the physiological role of p53 in exosome biology is accumulating, we are also learning more and more on the multifaceted role of mutant p53 in the regulation of exosomes and the resulting impact on cancer pathogenesis. Obviously, TP53 gene mutations compromise or ablate the effects of wild-type p53 on exosome biogenesis, as summarized above. In addition, similar as observed for the transcriptional activities of wild-type and mutant p53, mutant p53 proteins acquire novel exosomal functions that are distinct from those of wild-type p53, counteract wild-type p53-mediated tumor suppression, and typically promote tumor progression and metastatic spread. For example, p53-mutant colorectal tumor cells selectively shed miR-1246-enriched exosomes, even though intracellular miR-1246 levels are independent of p53 status [130]. In the model proposed, mutant p53 enhances SUMOylation of the RNA-binding protein hnRNPa2b1, which is required for binding and sorting miR-1246 into exosomes [130]. Uptake of miR-1246-enriched exosomes by neighboring macrophages triggers their reprogramming into a tumor-promoting M2-like state [130]. These tumor-associated macrophages (TAMs) are a chief component of the vast majority of solid tumors and strategic players in cancer progression, as they are responsible for constructing an immunosuppressive and pro-metastatic microenvironment through the production of chemokines, cytokines, and growth factors [130][131]. Although it is still unknown whether other stromal cells are also affected, this study revealed an intriguing cell nonautonomous pro-tumorigenic role of mutant p53 in microenvironmental reprogramming mediated by altered cargo loading of exosomes.
Similarly, a comparative proteomics analysis of p53-mutant and p53-null exosomes revealed a mutp53-associated reduction of podocalyxin (PODXL), a sialomucin associated with cancer aggressiveness [132][133]. In the proposed model, mutant p53 controls PODXL transcription via inhibition of p63 and Rab35 GTPase, which interacts with podocalyxin to influence its sorting to exosomes. When exosomes from p53-mutated cancer cells are taken up by fibroblasts, this promotes trafficking of integrins via the RCP/DGKα pathway. RCP-dependent integrin recycling is a well-characterized GOF mechanism of mutant p53 that increases tumor cell invasive migration in a cell-autonomous manner [76]. In addition, ECM proteins, also within tumors, are continuously assembled and remodeled, and integrin trafficking via the endosomal system can modulate the properties of the ECM [134][135]. Reprogramming of integrin trafficking in fibroblasts through the transfer of mutp53-exosomes therefore results in ECM remodeling and creation of pre-metastatic niches that invite metastatic invasion by tumor cells [133].
p53 mutants therefore elegantly modify secretion of extracellular vesicles in multiple ways to shape the cellular and extracellular microenvironment, both at the tumor site and in distant organs, to support invasive growth and metastatic spreading.
References
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263.
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52.
- Levine, A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989, 57, 1083–1093.
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078.
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370.
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758.
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431.
- Junttila, M.R.; Evan, G.I. p53—A Jack of all trades but master of none. Nat. Rev. Cancer 2009, 9, 821–829.
- Lu, W.J.; Amatruda, J.F.; Abrams, J.M. p53 ancestry: Gazing through an evolutionary lens. Nat. Rev. Cancer 2009, 9, 758–762.
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404.
- Levine, A.J.; Tomasini, R.; McKeon, F.D.; Mak, T.W.; Melino, G. The p53 family: Guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 259–265.
- Stiewe, T. The p53 family in differentiation and tumorigenesis. Nat. Rev. Cancer 2007, 7, 165–168.
- Bieging, K.T.; Attardi, L.D. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012, 22, 97–106.
- Jackson, J.G.; Lozano, G. The mutant p53 mouse as a pre-clinical model. Oncogene 2013, 32, 4325–4330.
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909.
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339.
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168.
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212.
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286.
- Stiewe, T.; Haran, T.E. How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist. Updates 2018, 38, 27–43.
- Tang, Q.; Su, Z.; Gu, W.; Rustgi, A.K. Mutant p53 on the Path to Metastasis. Trends Cancer 2020, 6, 62–73.
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317.
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30.
- Leroy, B.; Ballinger, M.L.; Baran-Marszak, F.; Bond, G.L.; Braithwaite, A.; Concin, N.; Donehower, L.A.; El-Deiry, W.S.; Fenaux, P.; Gaidano, G.; et al. Recommended Guidelines for Validation, Quality Control, and Reporting of TP53 Variants in Clinical Practice. Cancer Res. 2017, 77, 1250–1260.
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256.
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582.
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355.
- Sigal, A.R.; Rotter, V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res. 2005, 60, 6788–6793.
- Soussi, T.; Lozano, G. p53 mutation heterogeneity in cancer. Biochem. Biophys. Res. Commun. 2005, 331, 834–842.
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76.
- Klein, C.; Planker, E.; Diercks, T.; Kessler, H.; Kunkele, K.P.; Lang, K.; Hansen, S.; Schwaiger, M. NMR spectroscopy reveals the solution dimerization interface of p53 core domains bound to their consensus DNA. J. Biol. Chem. 2001, 276, 49020–49027.
- Dehner, A.; Klein, C.; Hansen, S.; Muller, L.; Buchner, J.; Schwaiger, M.; Kessler, H. Cooperative binding of p53 to DNA: Regulation by protein-protein interactions through a double salt bridge. Angew. Chem. Int. Ed. 2005, 44, 5247–5251.
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural basis of DNA recognition by p53 tetramers. Mol. Cell 2006, 22, 741–753.
- Kitayner, M.; Rozenberg, H.; Rohs, R.; Suad, O.; Rabinovich, D.; Honig, B.; Shakked, Z. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat. Struct. Mol. Biol. 2010, 17, 423–429.
- Weinberg, R.L.; Veprintsev, D.B.; Fersht, A.R. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004, 341, 1145–1159.
- Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429.
- Schlereth, K.; Beinoraviciute-Kellner, R.; Zeitlinger, M.K.; Bretz, A.C.; Sauer, M.; Charles, J.P.; Vogiatzi, F.; Leich, E.; Samans, B.; Eilers, M.; et al. DNA binding cooperativity of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2010, 38, 356–368.
- Schlereth, K.; Charles, J.P.; Bretz, A.C.; Stiewe, T. Life or death: p53-induced apoptosis requires DNA binding cooperativity. Cell Cycle 2010, 9, 4068–4076.
- Schlereth, K.; Heyl, C.; Krampitz, A.M.; Mernberger, M.; Finkernagel, F.; Scharfe, M.; Jarek, M.; Leich, E.; Rosenwald, A.; Stiewe, T. Characterization of the p53 cistrome--DNA binding cooperativity dissects p53’s tumor suppressor functions. PLoS Genet. 2013, 9, e1003726.
- Timofeev, O.; Schlereth, K.; Wanzel, M.; Braun, A.; Nieswandt, B.; Pagenstecher, A.; Rosenwald, A.; Elsasser, H.P.; Stiewe, T. p53 DNA binding cooperativity is essential for apoptosis and tumor suppression in vivo. Cell Rep. 2013, 3, 1512–1525.
- Timofeev, O.; Klimovich, B.; Schneikert, J.; Wanzel, M.; Pavlakis, E.; Noll, J.; Mutlu, S.; Elmshauser, S.; Nist, A.; Mernberger, M.; et al. Residual apoptotic activity of a tumorigenic p53 mutant improves cancer therapy responses. EMBO J. 2019, 38, e102096.
- Milner, J.; Medcalf, E.A. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 1991, 65, 765–774.
- Milner, J.; Medcalf, E.A.; Cook, A.C. Tumor suppressor p53: Analysis of wild-type and mutant p53 complexes. Mol. Cell. Biol. 1991, 11, 12–19.
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713.
- Shaulsky, G.; Goldfinger, N.; Rotter, V. Alterations in tumor development in vivo mediated by expression of wild type or mutant p53 proteins. Cancer Res 1991, 51, 5232–5237.
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46.
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872.
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860.
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580.
- Liu, D.P.; Song, H.; Xu, Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene 2010, 29, 949–956.
- Zhang, Y.; Xiong, S.; Liu, B.; Pant, V.; Celii, F.; Chau, G.; Elizondo-Fraire, A.C.; Yang, P.; You, M.J.; El-Naggar, A.K.; et al. Somatic Trp53 mutations differentially drive breast cancer and evolution of metastases. Nat. Commun. 2018, 9, 3953.
- Bougeard, G.; Sesboue, R.; Baert-Desurmont, S.; Vasseur, S.; Martin, C.; Tinat, J.; Brugieres, L.; Chompret, A.; de Paillerets, B.B.; Stoppa-Lyonnet, D.; et al. Molecular basis of the Li-Fraumeni syndrome: An update from the French LFS families. J. Med. Genet. 2008, 45, 535–538.
- Zerdoumi, Y.; Aury-Landas, J.; Bonaiti-Pellie, C.; Derambure, C.; Sesboue, R.; Renaux-Petel, M.; Frebourg, T.; Bougeard, G.; Flaman, J.M. Drastic effect of germline TP53 missense mutations in Li-Fraumeni patients. Hum. Mutat. 2013, 34, 453–461.
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604.
- Giacomelli, A.O.; Yang, X.; Lintner, R.E.; McFarland, J.M.; Duby, M.; Kim, J.; Howard, T.P.; Takeda, D.Y.; Ly, S.H.; Kim, E.; et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 2018, 50, 1381–1387.
- Turrell, F.K.; Kerr, E.M.; Gao, M.; Thorpe, H.; Doherty, G.J.; Cridge, J.; Shorthouse, D.; Speed, A.; Samarajiwa, S.; Hall, B.A.; et al. Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes Dev. 2017.
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000935.
- Gohler, T.; Jager, S.; Warnecke, G.; Yasuda, H.; Kim, E.; Deppert, W. Mutant p53 proteins bind DNA in a DNA structure-selective mode. Nucleic Acids Res. 2005, 33, 1087–1100.
- Brazdova, M.; Tichy, V.; Helma, R.; Bazantova, P.; Polaskova, A.; Krejci, A.; Petr, M.; Navratilova, L.; Ticha, O.; Nejedly, K.; et al. p53 Specifically Binds Triplex DNA In Vitro and in Cells. PLoS ONE 2016, 11, e0167439.
- Brazdova, M.; Navratilova, L.; Tichy, V.; Nemcova, K.; Lexa, M.; Hrstka, R.; Pecinka, P.; Adamik, M.; Vojtesek, B.; Palecek, E.; et al. Preferential binding of hot spot mutant p53 proteins to supercoiled DNA in vitro and in cells. PLoS ONE 2013, 8, e59567.
- Quante, T.; Otto, B.; Brazdova, M.; Kejnovska, I.; Deppert, W.; Tolstonog, G.V. Mutant p53 is a transcriptional co-factor that binds to G-rich regulatory regions of active genes and generates transcriptional plasticity. Cell Cycle 2012, 11, 3290–3303.
- Lozano, G. Restoring p53 in cancer: The promises and the challenges. J. Mol. Cell. Biol. 2019.
- Marin, M.C.; Jost, C.A.; Brooks, L.A.; Irwin, M.S.; O’Nions, J.; Tidy, J.A.; James, N.; McGregor, J.M.; Harwood, C.A.; Yulug, I.G.; et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat. Genet. 2000, 25, 47–54.
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887.
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295.
- Davison, T.S.; Vagner, C.; Kaghad, M.; Ayed, A.; Caput, D.; Arrowsmith, C.H. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J. Biol. Chem. 1999, 274, 18709–18714.
- Strano, S.; Fontemaggi, G.; Costanzo, A.; Rizzo, M.G.; Monti, O.; Baccarini, A.; Del Sal, G.; Levrero, M.; Sacchi, A.; Oren, M.; et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol. Chem. 2002, 277, 18817–18826.
- Wong, K.B.; DeDecker, B.S.; Freund, S.M.; Proctor, M.R.; Bycroft, M.; Fersht, A.R. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl. Acad. Sci. USA 1999, 96, 8438–8442.
- Li, Y.; Prives, C. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene 2007, 26, 2220–2225.
- Weisz, L.; Oren, M.; Rotter, V. Transcription regulation by mutant p53. Oncogene 2007, 26, 2202–2211.
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009, 137, 87–98.
- Neilsen, P.M.; Noll, J.E.; Mattiske, S.; Bracken, C.P.; Gregory, P.A.; Schulz, R.B.; Lim, S.P.; Kumar, R.; Suetani, R.J.; Goodall, G.J.; et al. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene 2013, 32, 2992–3000.
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218.
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341.
- Neilsen, P.M.; Noll, J.E.; Suetani, R.J.; Schulz, R.B.; Al-Ejeh, F.; Evdokiou, A.; Lane, D.P.; Callen, D.F. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget 2011, 2, 1203–1217.
- Shakya, R.; Tarulli, G.A.; Sheng, L.; Lokman, N.A.; Ricciardelli, C.; Pishas, K.I.; Selinger, C.I.; Kohonen-Corish, M.R.J.; Cooper, W.A.; Turner, A.G.; et al. Mutant p53 upregulates alpha-1 antitrypsin expression and promotes invasion in lung cancer. Oncogene 2017, 36, 4469–4480.
- Chang, Y.-T.; Lee, S.-H.; Liao, I.C.; Huang, S.-H.; Cheng, H.-C.; Liao, P.-C. Secretomic analysis identifies alpha-1 antitrypsin as required protein in cancer cell migration, invasion and pericellular fibronectin assembly for facilitating lung colonization of lung adenocarcinoma cells. Moll. Cell. Proteom. 2012, 11, 1320–1339.
- Kwon, C.H.; Park, H.J.; Choi, J.H.; Lee, J.R.; Kim, H.K.; Jo, H.J.; Kim, H.S.; Oh, N.; Song, G.A.; Park, D.Y. Snail and serpinA1 promote tumor progression and predict prognosis in colorectal cancer. Oncotarget 2015, 6, 20312–20326.
- Kwon, C.H.; Park, H.J.; Lee, J.R.; Kim, H.K.; Jeon, T.Y.; Jo, H.J.; Kim, D.H.; Kim, G.H.; Park, D.Y. Serpin peptidase inhibitor clade A member 1 is a biomarker of poor prognosis in gastric cancer. Br. J. Cancer 2014, 111, 1993–2002.
- El-Akawi, Z.J.; Al-Hindawi, F.K.; Bashir, N.A. Alpha-1 antitrypsin (alpha1-AT) plasma levels in lung, prostate and breast cancer patients. Neuroendocrinol. Lett. 2008, 29, 482–484.
- Taipale, J.; Lohi, J.; Saarinen, J.; Kovanen, P.T.; Keski-Oja, J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J. Biol. Chem. 1995, 270, 4689–4696.
- Frenzel, E.; Wrenger, S.; Brugger, B.; Salipalli, S.; Immenschuh, S.; Aggarwal, N.; Lichtinghagen, R.; Mahadeva, R.; Marcondes, A.M.; Dinarello, C.A.; et al. Alpha1-Antitrypsin Combines with Plasma Fatty Acids and Induces Angiopoietin-like Protein 4 Expression. J. Immunol. 2015, 195, 3605–3616.
- Weisz, L.; Damalas, A.; Liontos, M.; Karakaidos, P.; Fontemaggi, G.; Maor-Aloni, R.; Kalis, M.; Levrero, M.; Strano, S.; Gorgoulis, V.G.; et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 2007, 67, 2396–2401.
- Yeudall, W.A.; Vaughan, C.A.; Miyazaki, H.; Ramamoorthy, M.; Choi, M.Y.; Chapman, C.G.; Wang, H.; Black, E.; Bulysheva, A.A.; Deb, S.P.; et al. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis 2012, 33, 442–451.
- Martin, D.; Galisteo, R.; Gutkind, J.S. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J. Biol. Chem. 2009, 284, 6038–6042.
- Buganim, Y.; Solomon, H.; Rais, Y.; Kistner, D.; Nachmany, I.; Brait, M.; Madar, S.; Goldstein, I.; Kalo, E.; Adam, N.; et al. p53 Regulates the Ras circuit to inhibit the expression of a cancer-related gene signature by various molecular pathways. Cancer Res. 2010, 70, 2274–2284.
- Milyavsky, M.; Tabach, Y.; Shats, I.; Erez, N.; Cohen, Y.; Tang, X.; Kalis, M.; Kogan, I.; Buganim, Y.; Goldfinger, N.; et al. Transcriptional programs following genetic alterations in p53, INK4A, and H-Ras genes along defined stages of malignant transformation. Cancer Res. 2005, 65, 4530–4543.
- Solomon, H.; Buganim, Y.; Kogan-Sakin, I.; Pomeraniec, L.; Assia, Y.; Madar, S.; Goldstein, I.; Brosh, R.; Kalo, E.; Beatus, T.; et al. Various p53 mutant proteins differently regulate the Ras circuit to induce a cancer-related gene signature. J. Cell Sci. 2012, 125, 3144–3152.
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646.
- Dell’Orso, S.; Fontemaggi, G.; Stambolsky, P.; Goeman, F.; Voellenkle, C.; Levrero, M.; Strano, S.; Rotter, V.; Oren, M.; Blandino, G. ChIP-on-chip analysis of in vivo mutant p53 binding to selected gene promoters. OMICS 2011, 15, 305–312.
- Rahnamoun, H.; Lu, H.; Duttke, S.H.; Benner, C.; Glass, C.K.; Lauberth, S.M. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat. Commun. 2017, 8, 754.
- Yan, W.; Chen, X. Identification of GRO1 as a critical determinant for mutant p53 gain of function. J. Biol. Chem. 2009, 284, 12178–12187.
- Di Minin, G.; Bellazzo, A.; Dal Ferro, M.; Chiaruttini, G.; Nuzzo, S.; Bicciato, S.; Piazza, S.; Rami, D.; Bulla, R.; Sommaggio, R.; et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol. Cell 2014, 56, 617–629.
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314.
- Schwitalla, S.; Ziegler, P.K.; Horst, D.; Becker, V.; Kerle, I.; Begus-Nahrmann, Y.; Lechel, A.; Rudolph, K.L.; Langer, R.; Slotta-Huspenina, J.; et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013, 23, 93–106.
- Wong, G.S.; Lee, J.S.; Park, Y.Y.; Klein-Szanto, A.J.; Waldron, T.J.; Cukierman, E.; Herlyn, M.; Gimotty, P.; Nakagawa, H.; Rustgi, A.K. Periostin cooperates with mutant p53 to mediate invasion through the induction of STAT1 signaling in the esophageal tumor microenvironment. Oncogenesis 2013, 2, e59.
- Alford, A.I.; Hankenson, K.D. Matricellular proteins: Extracellular modulators of bone development, remodeling, and regeneration. Bone 2006, 38, 749–757.
- Vaughan, C.A.; Pearsall, I.; Singh, S.; Windle, B.; Deb, S.P.; Grossman, S.R.; Yeudall, W.A.; Deb, S. Addiction of lung cancer cells to GOF p53 is promoted by up-regulation of epidermal growth factor receptor through multiple contacts with p53 transactivation domain and promoter. Oncotarget 2016, 7, 12426–12446.
- Vogiatzi, F.; Brandt, D.T.; Schneikert, J.; Fuchs, J.; Grikscheit, K.; Wanzel, M.; Pavlakis, E.; Charles, J.P.; Timofeev, O.; Nist, A.; et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc. Natl. Acad. Sci. USA 2016, 113, E8433–E8442.
- Timofeev, O.; Stiewe, T. p53 gain-of-function mutations promote metastasis via ENTPD5 upregulation and enhanced N-glycoprotein folding. Mol. Cell. Oncol. 2017, 4, e1288678.
- Schneikert, J.; Stiewe, T. Pro-metastatic p53 mutants control folding of N-glycoproteins. Cell Cycle 2017, 16, 591–592.
- Fang, M.; Shen, Z.; Huang, S.; Zhao, L.; Chen, S.; Mak, T.W.; Wang, X. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 2010, 143, 711–724.
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049.
- Lamriben, L.; Graham, J.B.; Adams, B.M.; Hebert, D.N. N-Glycan-based ER Molecular Chaperone and Protein Quality Control System: The Calnexin Binding Cycle. Traffic 2016, 17, 308–326.
- Mackeen, M.M.; Almond, A.; Deschamps, M.; Cumpstey, I.; Fairbanks, A.J.; Tsang, C.; Rudd, P.M.; Butters, T.D.; Dwek, R.A.; Wormald, M.R. The conformational properties of the Glc3Man unit suggest conformational biasing within the chaperone-assisted glycoprotein folding pathway. J. Mol. Biol. 2009, 387, 335–347.
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211.
- Yu, L.; Weng, X.; Liang, P.; Dai, X.; Wu, X.; Xu, H.; Fang, M.; Fang, F.; Xu, Y. MRTF-A mediates LPS-induced pro-inflammatory transcription by interacting with the COMPASS complex. J. Cell Sci. 2014, 127, 4645–4657.
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; Le Quesne, J.; Baena Acevedo, J.D.; et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878.
- Pfister, N.T.; Fomin, V.; Regunath, K.; Zhou, J.Y.; Zhou, W.; Silwal-Pandit, L.; Freed-Pastor, W.A.; Laptenko, O.; Neo, S.P.; Bargonetti, J.; et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 2015, 29, 1298–1315.
- Ubertini, V.; Norelli, G.; D’Arcangelo, D.; Gurtner, A.; Cesareo, E.; Baldari, S.; Gentileschi, M.P.; Piaggio, G.; Nistico, P.; Soddu, S.; et al. Mutant p53 gains new function in promoting inflammatory signals by repression of the secreted interleukin-1 receptor antagonist. Oncogene 2015, 34, 2493–2504.
- Lee, S.; Margolin, K. Cytokines in cancer immunotherapy. Cancers 2011, 3, 3856–3893.
- Jain, A.; Kaczanowska, S.; Davila, E. IL-1 Receptor-Associated Kinase Signaling and Its Role in Inflammation, Cancer Progression, and Therapy Resistance. Front. Immunol. 2014, 5, 553.
- Schreuder, H.; Tardif, C.; Trump-Kallmeyer, S.; Soffientini, A.; Sarubbi, E.; Akeson, A.; Bowlin, T.; Yanofsky, S.; Barrett, R.W. A new cytokine-receptor binding mode revealed by the crystal structure of the IL-1 receptor with an antagonist. Nature 1997, 386, 194–200.
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67.
- Stetler-Stevenson, W.G. The role of matrix metalloproteinases in tumor invasion, metastasis, and angiogenesis. Surg. Oncol. Clin. 2001, 10, 383–392.
- Loging, W.T.; Reisman, D. Inhibition of the putative tumor suppressor gene TIMP-3 by tumor-derived p53 mutants and wild type p53. Oncogene 1999, 18, 7608–7615.
- Adissu, H.A.; McKerlie, C.; Di Grappa, M.; Waterhouse, P.; Xu, Q.; Fang, H.; Khokha, R.; Wood, G.A. Timp3 loss accelerates tumour invasion and increases prostate inflammation in a mouse model of prostate cancer. Prostate 2015, 75, 1831–1843.
- Martin del Campo, S.E.; Latchana, N.; Levine, K.M.; Grignol, V.P.; Fairchild, E.T.; Jaime-Ramirez, A.C.; Dao, T.V.; Karpa, V.I.; Carson, M.; Ganju, A.; et al. MiR-21 enhances melanoma invasiveness via inhibition of tissue inhibitor of metalloproteinases 3 expression: In vivo effects of MiR-21 inhibitor. PLoS ONE 2015, 10, e0115919.
- Haupt, S.; di Agostino, S.; Mizrahi, I.; Alsheich-Bartok, O.; Voorhoeve, M.; Damalas, A.; Blandino, G.; Haupt, Y. Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res. 2009, 69, 4818–4826.
- Haupt, S.; Raghu, D.; Haupt, Y. Mutant p53 Drives Cancer by Subverting Multiple Tumor Suppression Pathways. Front. Oncol. 2016, 6, 12.
- Haupt, S.; Mitchell, C.; Corneille, V.; Shortt, J.; Fox, S.; Pandolfi, P.P.; Castillo-Martin, M.; Bonal, D.M.; Cordon-Cardo, C.; Lozano, G.; et al. Loss of PML cooperates with mutant p53 to drive more aggressive cancers in a gender-dependent manner. Cell Cycle 2013, 12, 1722–1731.
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750.
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514.
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307.
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: A novel function of the p53 protein. Cancer Res. 2006, 66, 4795–4801.
- Yu, X.; Riley, T.; Levine, A.J. The regulation of the endosomal compartment by p53 the tumor suppressor gene. FEBS J. 2009, 276, 2201–2212.
- Schuldner, M.; Dorsam, B.; Shatnyeva, O.; Reiners, K.S.; Kubarenko, A.; Hansen, H.P.; Finkernagel, F.; Roth, K.; Theurich, S.; Nist, A.; et al. Exosome-dependent immune surveillance at the metastatic niche requires BAG6 and CBP/p300-dependent acetylation of p53. Theranostics 2019, 9, 6047–6062.
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771.
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51.
- Wang, J.; Zhao, Y.; Qi, R.; Zhu, X.; Huang, C.; Cheng, S.; Wang, S.; Qi, X. Prognostic role of podocalyxin-like protein expression in various cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 52457–52464.
- Novo, D.; Heath, N.; Mitchell, L.; Caligiuri, G.; MacFarlane, A.; Reijmer, D.; Charlton, L.; Knight, J.; Calka, M.; McGhee, E.; et al. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat. Commun. 2018, 9, 5069.
- Jones, M.C.; Caswell, P.T.; Moran-Jones, K.; Roberts, M.; Barry, S.T.; Gampel, A.; Mellor, H.; Norman, J.C. VEGFR1 (Flt1) regulates Rab4 recycling to control fibronectin polymerization and endothelial vessel branching. Traffic 2009, 10, 754–766.
- Shi, F.; Sottile, J. Caveolin-1-dependent beta1 integrin endocytosis is a critical regulator of fibronectin turnover. J. Cell Sci. 2008, 121, 2360–2371.
More
Information
Subjects:
Biology; Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
10 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No