 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | james duckers | + 1246 word(s) | 1246 | 2021-04-15 03:15:09 | | | |
| 2 | Karina Chen | -359 word(s) | 887 | 2021-06-09 07:45:50 | | | | |
| 3 | Karina Chen | -359 word(s) | 887 | 2021-06-09 07:46:19 | | | | |
| 4 | Karina Chen | -359 word(s) | 887 | 2021-06-09 07:46:38 | | |
Video Upload Options
Cystic fibrosis (CF) is a rare, progressive, multi-organ genetic disease. CF results from mutations in the CF transmembrane conductance regulator (CFTR) gene, which lead to defective transport of chloride and sodium ions across the epithelial cell membrane of multiple organs, including the lungs, pancreas, gallbladder, intestine, and reproductive system. The recently developed CFTR modulators treat the underlying cause of CF. Ivacaftor (Kalydeco®, Vertex Pharmaceuticals Incorporated, Boston, MA, USA), a small-molecule CFTR modulator, was the first approved CFTR modulator. Although clinical trials have provided valuable evidence on the safety and efficacy of ivacaftor, it is important that real-world outcomes in routine clinical practice are understood. Here, we systematically reviewed data from real-world observational studies of ivacaftor treatment in people with CF.
1. Introduction
Cystic fibrosis (CF) is a rare, multi-organ genetic disease with a median age at death of 30.8 years in the US (2018) and 29.0 years in Europe (2017), and it affects an estimated 70,000 to 100,000 people worldwide.[1][2][3] Mutations in the CF transmembrane conductance regulator (CFTR) gene affect CFTR protein expression and/or function at the epithelial cell surface.[4] This results in defective transport of chloride and sodium ions across the epithelial cell membrane of multiple organs, such as the lungs, pancreas, gallbladder, and intestine.5 People with CF (pwCF) can experience declining lung function and pulmonary exacerbations, and the primary cause of death for pwCF is respiratory failure.[5] In addition, extrapulmonary complications can occur, which include exocrine pancreatic damage, gastrointestinal dysfunction leading to poor nutrition and growth, and damage to the endocrine pancreas over time, resulting in the development of CF-related diabetes.[6][7][8][9][10] These comorbidities result in high levels of healthcare utilization and have been linked to morbidity and mortality.[9]
The recent development of CFTR modulators that address the underlying cause of CF has the potential to change the disease trajectory. Ivacaftor (Kalydeco®, Vertex Pharmaceuticals Incorporated, Boston, MA, USA), a CFTR potentiator, was the first approved CFTR modulator therapy for pwCF aged ≥6 years with a G551D CFTR mutation.[11][12] Following initial approval, ivacaftor has been granted a series of label expansions by the US Food and Drug Administration and the European Medicines Agency to include additional CFTR mutations in pwCF as young as 4 months of age.[13][14]
While clinical trial data provide valuable evidence on safety and efficacy, it is important that we understand the real-world outcomes associated with ivacaftor in routine clinical practice. We have, therefore, conducted and published a systematic review of the evidence available for pwCF treated with ivacaftor from numerous large registries and smaller studies investigating the safety and effectiveness of ivacaftor in the real-world setting.[15]
2. Methods
A literature review was conducted based on a predefined search and selection protocol, with search criteria including studies published as journal articles or conference abstracts enrolling six or more pwCF treated with ivacaftor (January 1, 2012 to September 4, 2019) in MEDLINE or Embase (searched via ProQuest), and studies written in the English language. Clinical case reports as well as interventional (randomized or non-randomized), animal, and preclinical studies were excluded.
Key outcomes were prespecified by clinical experts to include those related to lung function, pulmonary exacerbations, nutritional parameters (e.g., body mass index and weight), endocrine and exocrine pancreatic functions (e.g., CF-related diabetes and pancreatic insufficiency), healthcare utilization, lung microbiome, patient-reported outcomes (e.g., Cystic Fibrosis Questionnaire-Revised [CFQ-R] respiratory symptom scale and physical function tests [e.g., 6-minute walk test]), safety, CF-related complications, organ transplantation, and mortality.
3. Results
We identified 368 publications of real-world studies (Supplementary Figure 1). Overall, 75 publications, comprising 57 unique studies, met the inclusion criteria.
Studies reporting within-group changes for pwCF treated with ivacaftor showed consistent improvements in lung function observed as early as 1 month following ivacaftor treatment.
Increases in weight and body mass index were consistently observed after ivacaftor was initiated and improvements continued over time. There were decreases in pulmonary exacerbations, healthcare resource utilization such as hospitalizations, antibiotic use, and antibiotic duration, and Pseudomonas aeruginosa prevalence reported for up to 66 months post-ivacaftor treatment. In addition, improvements in patient-reported outcomes, such as the CFQ-R and the Sino-Nasal Outcome Test, were also observed following ivacaftor treatment. Similar clinical benefits were seen in studies comparing ivacaftor to modulator untreated comparators, in addition to decreases in mortality, organ transplantation, and CF-related complications. Furthermore, no new safety concerns were identified for ivacaftor during this review.
In summary, the improvement in outcomes associated with real-world ivacaftor use summarized in this study demonstrate the substantial benefit of CFTR modulation with ivacaftor on pulmonary and non-pulmonary outcomes, many of which were not studied in clinical trials.
4. Conclusions
This systematic literature review of real-world studies, encompassing a range of study designs and data sources, geographic settings, patient characteristics, and study durations, consistently identified improvements in a broad range of outcomes for pwCF receiving ivacaftor. The real-world evidence collectively supports the value of ivacaftor as a long-term treatment that can fundamentally modify the progression of CF by treating the underlying cause of the disease, and potentially reduce disease burden, while allowing pwCF to have better survival outcomes and improved quality of life.
Supplementary material
Supplementary Figure 1. Identification of publications according to PRISMA guidelines
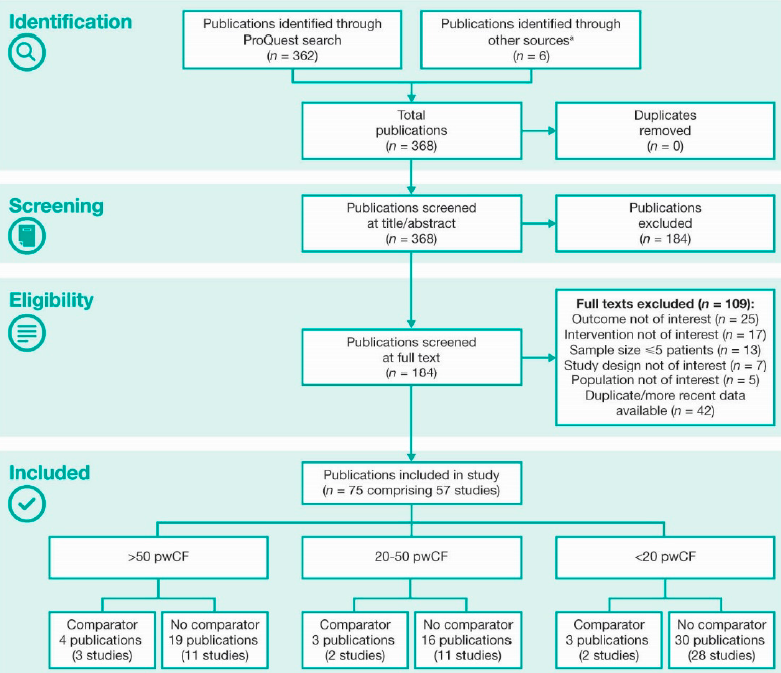 pwCF, people with CF
pwCF, people with CF
Funding: The study was funded by Vertex Pharmaceuticals Incorporated.
Acknowledgments: Medical writing assistance was provided by Rob Kite, BSc (Hons), of Complete HealthVizion, who was contracted and compensated by Vertex Pharmaceuticals Incorporated.
References
- Cystic Fibrosis Foundation. 2018 Patient Registry: Annual Data Report. 2019. Available at: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2018-Patient-Registry-Annual-Data-Report.pdf. Accessed January 2, 2020.
- European Cystic Fibrosis Society. ECFS Patient Registry: Annual Data Report. 2017. Available at: https://www.ecfs.eu/sites/default/files/general-content-images/working-groups/ecfs-patient-registry/ECFSPR_Report2017_v1.3.pdf. Accessed July 9, 2020.
- Kelly J. Environmental scan of cystic fibrosis research worldwide. J Cyst Fibros 2017;16:367–370.
- Brown SD, White R, Tobin P. Keep them breathing: Cystic fibrosis pathophysiology, diagnosis, and treatment. JAAPA 2017;30:23–27.
- O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet 2009;373:1891–1904.
- Hassan M, Bonafede MM, Limone BL, et al. The burden of cystic fibrosis in the Medicaid population. Clinicoecon Outcomes Res 2018;10:423–431.
- Peterson ML, Jacobs DR, Jr., Milla CE. Longitudinal changes in growth parameters are correlated with changes in pulmonary function in children with cystic fibrosis. Pediatrics 2003;112:588–592.
- Zemel BS, Jawad AF, FitzSimmons S, et al. Longitudinal relationship among growth, nutritional status, and pulmonary function in children with cystic fibrosis: Analysis of the Cystic Fibrosis Foundation National CF Patient Registry. J Pediatr 2000;137:374–380.
- Brennan AL, Beynon J. Clinical updates in cystic fibrosis-related diabetes. Semin Respir Crit Care Med 2015;36:236–250.
- Hassan M, Bonafede M, Limone B, et al. The burden of cystic fibrosis: Pulmonary exacerbations and healthcare resource utilization in a commercially insured US population. Poster presented at the ISPOR 21st Annual International Meeting, Washington, DC, USA, May 21–25, 2016.
- Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med 2013;187:1219–1225.
- Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011;365:1663–1672.
- Vertex Pharmaceuticals (Europe) Limited. Kalydeco 150 mg film-coated tablets SmPC. 2020. Available at: https://www.medicines.org.uk/emc/product/3040/smpc. Accessed December 17, 2020.
- Vertex Pharmaceuticals Incorporated. Kalydeco PI. 2020. Available at: https://www.drugs.com/pro/kalydeco.html. Accessed December 17, 2020.
- Duckers J, Lesher B, Thorat T, et al. Real-world outcomes of ivacaftor treatment in people with cystic fibrosis: A systematic review. J Clin Med 2021;10:1527.


