Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Javier Molina-Cerrillo | + 3253 word(s) | 3253 | 2021-06-01 05:42:23 | | | |
| 2 | Peter Tang | Meta information modification | 3253 | 2021-06-08 05:32:38 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Molina-Cerrillo, J. Targeting Tyrosine Kinases in ccRCC. Encyclopedia. Available online: https://encyclopedia.pub/entry/10598 (accessed on 08 February 2026).
Molina-Cerrillo J. Targeting Tyrosine Kinases in ccRCC. Encyclopedia. Available at: https://encyclopedia.pub/entry/10598. Accessed February 08, 2026.
Molina-Cerrillo, Javier. "Targeting Tyrosine Kinases in ccRCC" Encyclopedia, https://encyclopedia.pub/entry/10598 (accessed February 08, 2026).
Molina-Cerrillo, J. (2021, June 07). Targeting Tyrosine Kinases in ccRCC. In Encyclopedia. https://encyclopedia.pub/entry/10598
Molina-Cerrillo, Javier. "Targeting Tyrosine Kinases in ccRCC." Encyclopedia. Web. 07 June, 2021.
Copy Citation
Clear cell renal cell carcinoma (ccRCC) is the seventh most frequently diagnosed tumor in adults in Europe and represents approximately 2.5% of cancer deaths. Currently, there are multiple therapeutic drugs available for advanced disease, including therapies against VEGFR with successful results in patients´ survival. Other tyrosine kinases’ pathways, including PDGFR, Axl or MET have emerged as key signaling pathways involved in RCC biology.
Tyrosine kinase
Kidney cancer
Vascular endothelial growth factor receptor (VEGFR)
Platelet Derived Growth Factor Receptor (PDGFR)
Tyrosine-Protein Kinase Met (MET)
Axl
Fibroblast Growth factor Receptor (FGFR)
1. Introduction
Kidney cancer represents the third tumor of the urinary tract most frequently diagnosed in adults. In Europe, the incidence of clear cell renal cell carcinoma (ccRCC) accounts for 84,000 new cases per year with a mortality rate of about 35,000 patients in 2012 [1]. The clear cell subtype is the most common one representing approximately 85% to 90% of all renal cancer diagnosis [2]. The mortality rate varies significantly throughout different regions and is associated to the availability of more sophisticated diagnostic techniques, multidisciplinary teams and effective systemic drugs. In fact, in countries from Northern and Eastern Europe, Australia and US, the mortality tends to stabilize or even decline [3]. Additionally, the incidence of kidney cancer is greater in men than women (ratio of 2:1) [4] and it is usually diagnosed around the sixth decade of life (median age around 67 years).
In recent years, kidney cancer diagnosis has increased among early stages and only 20% of patients present metastases at tumor diagnosis. Despite radical treatment for localized disease, around 20% to 40% of patients relapse within five years [5][6].
Until 2006, the treatment of the metastatic disease was based on cytokines that had an unfavorable safety profile and achieved poor efficacy results with a progression-free survival (PFS) of around five months and overall survival (OS) of around 21 months [7].
The initial discovery of the role that angiogenesis has in kidney cancer development and progression has led the research of new agents that target key players of this step. The efficacy demonstrated by sunitinib, a tyrosine kinase inhibitor (TKI), targeting mainly the vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor (PDGFR), has been able to demonstrate a change in the natural history of metastatic kidney cancer and, quickly, prompted the use of TKI in the therapeutic algorithm of metastatic ccRCC patients. Sunitinib in the first line setting achieved an increase in the median PFS to up to 11 months and OS to approximately 26 months [8].
Since then and until now, more drugs have been developed and approved by the regulatory agencies for the treatment of metastatic ccRCC including tyrosine kinase inhibition strategies alone or in combination with other therapeutic approaches. For example, the inhibition of the mTORC complex by everolimus or temsirolimus presented benefits in a selected patients´ population and new immunotherapy strategies, such as nivolumab or its combination with ipilimumab, presented positive results in terms of efficacy and survival post VEGF-therapy and in the first line setting respectively [9][10].
2. Molecular Biology of Kidney Cancer
There are several genes involved in the development of both sporadic and hereditary renal cancer. Among them, VHL is the most important gene, standing at around 50% of ccRCC [3]. Mutations in the PTEN/PI3K/mTOR axis lead to permanent activation of the mTORC complex, underlying ccRCC development and progression.
Epigenetic alterations are also critical for ccRCC development, therefore genes involved in chromatin remodeling, such as PBRM1, BAP1, SETD2, and KDM5C, also play a role in this setting of the disease.
These gene alterations and the phenotypic results within the biology of kidney cancer cells are key to understanding the exact mechanisms responsible for ccRCC development and progression and the underlying mode of action of the treatment used for these patients in daily practice [11][12].
3. Tyrosine Kinases and Coupled Intracellular Signaling Involved in RCC
3.1. Vascular Endothelial Growth factor Receptor (VEGFR)
Vascular endothelial growth factor receptors (Figure 1) represent a family of three receptors: VEGFR1, VEGFR2 and VEGFR3. The most important one, from a biologically point of view, is VEGFR2, which is the main receptor for VEGF in the vascular endothelium, regulating different intracellular processes [13].
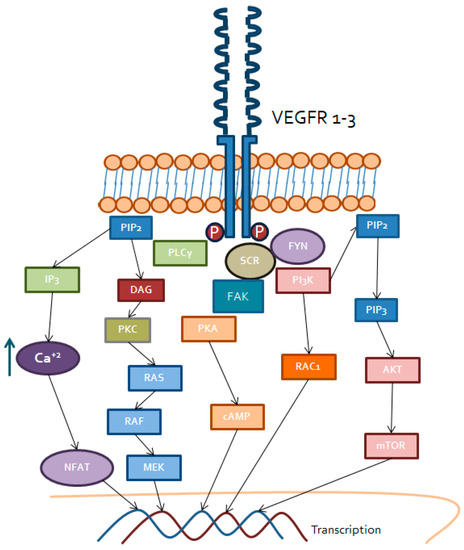
Figure 1. Intracellular signaling triggered by VEGF receptors. Specific transmembrane receptor tyrosine kinase for VEGF, i.e., VEGFR 1–3, recruit PLCγ, Src and FYN in order to signal mainly through DAG receptors (PKC), PKA, Rac1, MEK and Akt/mTOR. Transcription factors activated by these signaling, including NFAT or AP1, will finally regulate gene expression associated to VEGFR/VEGF binding. Increased levels of intracellular calcium also linked to this binding will contribute to PKC and NFAT activation, among others.
All members of the VEGFR family have the same structure, consisting in a seven extracellular immunoglobulin-like domain, a transmembrane domain and an intracellular tyrosine kinase domain. They are mainly regulated by their soluble ligands, among which are VEGF A/B/C/D, placenta growth factor (PlGF), parapoxvirus VEGFE, snake venom VEGFF and neuropilins NRP1 and NRP2 [14][15]. However, there is another way of VEGFR activation, mainly VEGFR2, by non-canonical activation mechanisms that would explain other functions that are attributed to this receptor. These non-canonical ways of activation would include mechanical stimulation, with the formation of mechanosensory complexes between VEGFR2 or vascular endothelial cadherin and platelet endothelial cell adhesion molecule 1 (PECAM1). When the complex is built, it is able to regulate the endothelial nitric oxide synthase (eNOS) [14][16]. In addition, VEGFR2 can interact with galectins, mainly galectin 3, expressed ubiquitously on the cell surface and with low density lipoproteins (LDL) [17].
As we have already presented, the VEGF ligands are able to bind their membrane receptors, but with different affinities. VEGFA is able to bind all three receptors, VEGFC and D do the same, but mainly to VEGFR2 and VEGFR3, and VEGFB and PIGF have greater affinity for VEGFR1 binding [18][19].
Once the ligand is bound, stable VEGFR dimers are generated to trigger activation of the tyrosine kinase domains and initiate intracellular signaling. The most important dimers in the vascular endothelium are the VEGFR2 homodimers. However, heterodimers between VEGFR1-VEGFR2 and VEGFR2-VEGFR3 can also be present. The first one exists in atherosclerotic lesions and in the embryonic endothelium and the second one, mainly in the lymphatic endothelium. Those heterodimers are known to be involved in cancer-related processes, but by not yet clearly defined paths [14][20]. Intracellular signaling triggered involves PI3K-AKT-mTOR and phospholipase Cγ (PLCγ) pathways.
3.2. Platelet Derived Growth Factor Receptor (PDGFR)
Platelet derived growth factor receptors consist of two members: PDGFRα and PDGFRβ. These receptors are composed of five extracellular immunoglobulin-like domains, a transmembrane domain and an intracellular tyrosine kinase domain in the C-terminal region [21]. Their signaling is fundamental in embryonic development, as well as in wound repair. Moreover, there is an increased interest about their involvement in pathological processes, such as cancer, due to their role in tumor microenvironment maintenance [22].
For their tyrosine kinase domain activation, these receptors need a ligand binding to achieve subsequent stabilization and dimerization. PDGFs are synthesized as pre-proteins and require the activity of proteases to expose their mature and active form. There are four dimeric isoforms, as PDGF homodimer: PDGF AA/BB/CC/DD and one heterodimeric isoform constituted by PDGF AB [22].
PDGFs are synthesized by different cell types, mainly mesenchymal cells, endothelial cells, some epithelial strains and myeloid cells, such as macrophages or platelets. Moreover, all these cells also express PDGFRα and/or PDGFRβ, so that PDGFs usually lead to autocrine regulation loops [23][24].
Similar to other tyrosine kinase receptors, not all PDGF subtypes result in the formation of the same dimers. PDGF-BB, PDGF-CC, PDGF-DD and PDGF-AB lead to the formation of the dimers PDGFRα/PDGFRβ, PDGF-AA, PDGF-AB, PDGF-BB and PDGF-CC do the same with the dimers PDGFRα/PDGFRα and PDGF-BB and PDGF-DD lead to PDGFRβ/PDGFRβ [25][26].
Once the intracellular tyrosine kinase domain is activated, PDGFR is able to regulate several signaling pathways. On the one hand, PI3K is attracted to the receptor and later activated. PI3K activation transforms PIP2 into PIP3, which is able to bind and activate AKT and, therefore, trigger the mTOR complex to regulate genes related to cell survival [27]. In addition, the tyrosine kinase domain is also able to attract PLCγ, which transforms PIP2 into IP3 and DAG, increasing intracellular calcium and activating PKC to ultimately regulate cell growth and survival [28]. On the other hand, after activation of the tyrosine kinase domain, an interaction with the Nck adapter protein occurs and C-Jun N-terminal protein-serine/threonine kinase (JNK) is activated by the RAS and MAPK pathway. Cell proliferation and survival processes are consequently regulated after PDGFR activation [29].
Active PDGFR is also able to recruit Src and, together with Gbr2 and Sos, is able to phosphorylate RAS-GDP to RAS-GTP. Consequently, there is an activation of relevant oncogenic signaling pathways via RAS/RAF/ERK, which mainly regulate cell proliferation-related genes [30].
It should be noted that PDGFR is intimately linked and related to KIT and FLT3, which explains why TKIs with activity against all these targets are effective in the treatment of renal cancer [31].
3.3. Tyrosine-Protein Kinase Met (MET)
Tyrosine-protein kinase MET (Figure 2) or the hepatocyte growth factor receptor (HGFR) is a membrane receptor involved in multiple biological routes. Initially, it is synthesized as a single chain and, subsequently, through post-transcriptional modulation, the α and β subunit result in the mature receptor as a disulfide-linked heterodimer [32].
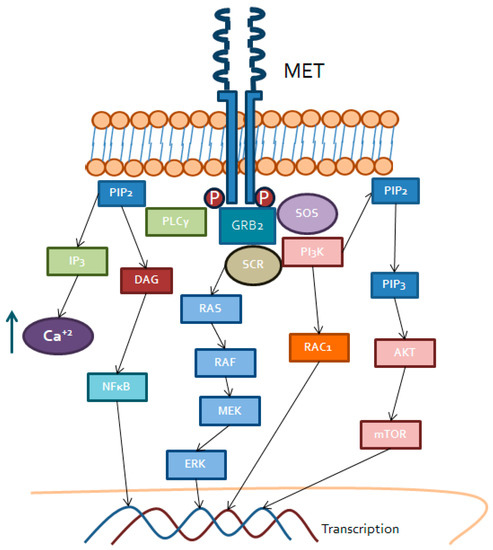
Figure 2. MET triggered intracellular signaling. Binding of HGF to MET transmembrane receptor tyrosine kinase recruit PLCγ, GRB2 and SOS, which activate DAG receptors (PKC), RAS/RAF/MEK, PI3K/RAC and Akt/mTOR. These kinases lead to activation of transcription factors including NFκB or AP1 for gene expression.
MET has an extracellular domain with four subdomains: immunoglobulin-like for ligand binding, a membrane anchoring domain and an intracellular domain, at the C-terminal end of the protein where the tyrosine kinase region is located, with different binding sites for substrates and ligands [33].
The known ligand of MET is the hepatocyte growth factor (HGF). Mainly by mesenchymal cells, it is secreted into the medium as an inactive form and, by the activity of extracellular proteases, it turns into a mature protein. At this time, HGF is able to perform its paracrine function on epithelial cells where it is usually expressed [34]. HGF regulates cell proliferation, motility, survival and growth functions of endothelial cells and also the hydrolysis of extracellular matrix proteins. The latest function justifies the key role of MET in embryonic development, as well as in other pathological processes, such as cancer [35].
Once HGF binds MET, it produces oligomerization of the receptor and the subsequent activation of the tyrosine kinase domain to exert the transcriptional regulation [36]. The expression of HGF and MET, as well as the regulation of this pathway, can also be affected by interleukins, such as IL-1and IL-6 or hypoxia-related factors, such as HIF 1α/2α, tumor necrosis factor-α (TNFα) or some subtypes of FGF, such as bFGF [37].
Once the tyrosine kinase domain is activated, several molecular signaling pathways are stimulated. Those pathways are mainly PI3K/AKT and RAS/MAPK/ERK together with other auxiliary proteins, such as Sos, Gbr2 and including the activation of Src, along with NFκB regulation [38][39]. All these pathways regulate, therefore, cell proliferation, survival, angiogenesis, motility and epithelial to mesenchymal transition (EMT).
The HGF/MET pathway interacts with other cell pathways that can reinforce its activation. In fact, in this sense, several escape mechanisms to various drugs have been described. Interaction with EGFR/HER2 mainly occurs because EGFR can activate Src-dependent MET in a ligand-independent manner [35]. HGF/MET also interacts with the VEGFR2 pathway, mainly by increasing VEFGA, and the NOTCH or βcatenin and WNT pathways, which partly explains the role of this receptor in EMT, angiogenesis and tumor growth and migration [40][41].
3.4. Axl
Axl is part of the TAM receptors family, constituted by three members: Tyro3, Mer and Axl itself. The TAM family is expressed mainly in the liver, central nervous system, platelets and in some cells of the innate immune system and the vascular endothelium [42]. All of them have different physiological functions, but they have in common their important relationship with cell proliferation and motility in cancer cells. Axl is also involved in angiogenesis and hematopoiesis, unlike Mer that is involved in immunosuppressive cell mechanisms [43][44].
The Axl structure is similar to other tyrosine kinase receptors. It has an extracellular domain with two immunoglobulin-like ligand binding domains, a transmembrane region and an intracellular region where the tyrosine kinase dominion is located [45].
Growth arrest-specific protein 6 (Gas6) is the most important ligand of Axl, but it is not the only one, since tubby-like protein 1 (TULP-1) can also enhance the activation of this receptor. Usually, the binding of Gas6 with Axl occurs with two molecules of Gas6 favoring the dimerization of two Axl proteins, with the subsequent autophosphorylation of the tyrosine residues in the tyrosine kinase domain and the activation of the dependent intracellular signaling cascades [43][46].
Although the most frequent way for Axl activation is through Gas6, there are other alternative manners. Axl can also be activated independently from the receptor: Under oxidative stress conditions, by interaction with another Axl receptor in the same cell or with the one of a nearby cell or by hetero-dimerization with other receptor families, such as VEGFR1 or other members of the TAM family [47].
Once tyrosine kinase domain is activated, Axl is able to exert its activity through different signaling pathways. It is able to recruit PI3K/AKT/mTOR that regulates cell survival through the expression of NFκB. Furthermore, it acts over the RAS/RAF/ERK and PI3K/RAC pathways to regulate proliferation and cell motility. In addition, it affects other membrane receptors such as MET, VEGFR or EGFR and participates in EMT processes promoting invasiveness [48][49].
3.5. Fibroblast Growth factor Receptor (FGFR)
FGFR is a family of membrane receptors consisting of five components [53]. FGFR1-4 has tyrosine kinase activity, while FGFR5, although it seems to have affinity for the FGFs, has no intracellular tyrosine kinase domain [54].
This family of receptors binds to FGF ligands that are secreted into the medium in order to be activated and fulfill their intracellular functions. The FGF ligands are divided into seven subfamilies according to their origin and structure [55][56][57]:
-
FGF1: Constituted by FGF1, is also known as aFGF or FGF acid and FGF2 or bFGF (basic FGF);
-
FGF4: Constituted by FGF4, FGF5 and FGF6;
-
FGF7: Constituted by FGF3, FG7, FGF10 and FGF22;
-
FGF9: Constituted by FGF9, FGF16 and FGF20;
-
FGF8: Constituted by FGF8, FGF17 and FGF18.
These forms are considered canonical or paracrine and recruit heparin or heparan sulfate as a cofactor to activate FGFR.
-
FGF11: Constituted by FGF11, FGF12, FGF13 and FGF14.
The FGF11 family is also known as intracellular FGFs and use ion channels as activating cofactors.
-
FGF15/19: Constituted by FGF15/19, FGF21 and FGF23.
This subfamily is also known as the endocrine FGFs and they use proteins from the Klotho family as cofactors: FGF 15/19 and FGF21 mainly need βKlotho and FGF23 needs αKlotho.
All FGFs are not expressed in the same spatial or temporal framework, in fact, some of them are only relevant in embryonic development [58]. However, others play an important role in adult life: Activating FGFRs. These are expressed by different types of tissues and regulate cell proliferation, migration, survival and differentiation. In addition, they also regulate other growth factor receptors such as EGFR, VEGF or HGF, so a role in angiogenesis and inflammation has been suggested for FGFR [59][60].
FGFR exhibits a structure including three immunoglobulin-like extracellular domains that bind to FGFs, a transmembrane domain and an intracellular tyrosine kinase domain [61]. Once FGF binds, FGFR dimerizes causing autophosphorylation of the tyrosine residues in the tyrosine kinase domain and triggers the intracellular signaling cascade. FGFR is able to recruit Src and its effectors, to finally activate RAS/ERK and also, directly phosphorylates FGFR substrate 2 (FRS2) that is able to activate the PI3K/AKT pathway. In addition, the activation of FGFR leads the activation of PLCγ and PKC to activate MAPK [57][62][63].
4. The Impact of Immunotherapy in an Angiogenic Disease
The involvement of VHL in kidney cancer was the first hit that allowed the development of all the previously mentioned TKIs treatments leading a historic change in metastatic RCC survival. Moreover, the immune system has also been investigated as a relevant player in kidney cancer behaviour and target agents have demonstrated survival impact in clinical trials with novel immune-based therapies. Indeed, the benefit in OS was reached by the Checkmate 025 trial where patients in the second- and third-line treatment were randomized to receive nivolumab versus everolimus [64]. The primary endpoint was achieved (median OS = 25 months for nivolumab versus 19.6 months for everolimus (HR = 0.73, p = 0.002)) and nivolumab became a new standard of care after treatment with a VEGFR inhibitor. Furthermore, the double immunotherapy combination (PD1/CTLA4) was evaluated in the first line setting in the Checkmate 214 trial including 1096 patients that were randomized to receive nivolumab 3 mg/kg plus ipilimumab 1 mg/kg versus sunitinib at standard dose [10]. The results showed a significant benefit in the coprimary endpoints of ORR (42% with nivolumab plus ipilimumab versus 27% with sunitinib, p < 0.001), median PFS (11.6 months with nivolumab plus ipilimumab and 8.4 months with sunitinib, HR = 0.82, p = 0.03) and median OS (not reached with nivolumab plus ipilimumab versus 26.0 months with sunitinib, HR = 0.63, p < 0.001), among intermediate- and poor-risk patients. After these results, the double immunotherapy combination was approved in this setting. Even though it was not considered a primary endpoint of the trial, interesting results were reported from the favorable risk MSKCC group. In this exploratory analysis, the ORR was 29% (11% of complete responses) with nivolumab plus ipilimumab versus 52% (6% of complete responses) with sunitinib. The median PFS in this subgroup was 15.3 months with nivolumab plus ipilimumab versus 25.1 months with sunitinib and the median OS was not reached with nivolumab plus ipilimumab compared with 32.9 months in the sunitinib arm. The updated results of the coprimary endpoints presented in the 2019 Genitourinary Cancers Symposium after a minimum follow up of 30 months (median 32.4 months) still show a benefit of nivolumab plus ipilimumab in the Intention-To-Treat (ITT) and intermediate- and poor-risk patients [65]. On the contrary, in the favourable-risk group, no significant benefit was reported.
Remarkably, immunotherapy has also been combined with TKIs in the first line setting of kidney cancer. This treatment strategy is currently being evaluated in different phase III trials, two of them have recently published their results [66][67]. The first one is the MK426 trial that randomized 861 patients with metastatic ccRCC to receive treatment with pembrolizumab 200 mg plus axitinib 5 mg/12 h versus sunitinib 50 mg/24 h four weeks on/two weeks off [66]. After a median follow-up of 12.8 months, the study met its primary endpoints with a median OS not reached in both groups (HR = 0.53; p < 0.0001) and a median PFS of 15.1 months with pembrolizumab plus axitinib versus 11.1 months with sunitinib (HR = 0.69; p < 0.001). The ORR was 59.3% (complete responses = 5.8%) with pembrolizumab plus axitinib and 35.7% (complete responses = 1.9%) with sunitinib. The second trial is the JAVELIN RENAL 101 that randomized 886 patients to receive avelumab 10 mg/kg plus axitinib 5 mg/12 h versus sunitinib 50 mg/24 h four weeks on/two weeks off [67]. The study also met its coprimary endpoints of PFS and OS among patients with PDL-1 positive tumors. A PDL1-positive tumor was found in 560 patients (63.2%) and, after a median follow up of 11.6 months, the median PFS was 13.8 months with avelumab plus axitinib versus 7.2 months with sunitinib (HR = 0.61; p < 0.001) and data on OS were still immature due to the low number of events (37 patients had died in the avelumab plus axitinib group and 44 patients had died in the sunitinib group).
References
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403.
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009.
- Znaor, A.; Lortet-Tieulent, J.; Laversanne, M.; Jemal, A.; Bray, F. International variations and trends in renal cell carcinoma incidence and mortality. Eur. Urol. 2015, 67, 519–530.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Dabestani, S.; Thorstenson, A.; Lindblad, P.; Harmenberg, U.; Ljungberg, B.; Lundstam, S. Renal cell carcinoma recurrences and metastases in primary on-metastatic patients: A population-based study. World J. Urol. 2016, 34, 1081–1086.
- Kane, C.J.; Mallin, K.; Ritchey, J.; Cooperberg, M.R.; Carroll, P.R. Renal cell cancer stage migration: Analysis of the National Cancer Data Base. Cancer 2008, 113, 78–83.
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus Interferon Alfa in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 115–124.
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Oudard, S.; Negrier, S.; Szczylik, C.; Pili, R.; Bjarnason, G.A.; et al. Overall Survival and Updated Results for Sunitinib Compared with Interferon Alfa in Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2009, 27, 3584–3590.
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456.
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290.
- Nickerson, M.L.; Jaeger, E.; Shi, Y.; Durocher, J.A.; Mahurkar, S.; Zaridze, D.; Matveev, V.; Janout, V.; Kollarova, H.; Bencko, V.; et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin. Cancer Res. 2008, 14, 4726–4734.
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.-M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90.
- Parveen, A.; Subedi, L.; Kim, H.W.; Khan, Z.; Zahra, Z.; Farooqi, M.Q.; Kim, S.Y. Phytochemicals Targeting VEGF and VEGF-Related Multifactors as Anticancer Therapy. J. Clin. Med. 2019, 8, 350.
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625.
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584.
- Wang, S.; Iring, A.; Strilic, B.; Albarrán-Juárez, J.; Kaur, H.; Troidl, K.; Tonack, S.; Burbiel, J.C.; Müller, C.E.; Fleming, I.; et al. P2Y2 and Gq/G11 control blood pressure by mediating endothelial mechanotransduction. J. Clin. Investig. 2015, 125, 3077–3086.
- Dalziel, M.; Crispin, M.; Scanlan, C.N.; Zitzmann, N.; Dwek, R.A. Emerging principles for the therapeutic exploitation of glycosylation. Science 2014, 343, 1235681.
- Sarabipour, S.; Ballmer-Hofer, K.; Hristova, K. VEGFR-2 conformational switch in response to ligand binding. Elife 2016, 5, e13876.
- Anisimov, A.; Leppänen, V.M.; Tvorogov, D.; Zarkada, G.; Jeltsch, M.; Holopainen, T.; Kaijalainen, S.; Alitalo, K. The basis for the distinct biological activities of vascular endothelial growth factor eceptor-1 ligands. Sci. Signal. 2013, 6, ra52.
- Deng, Y.; Zhang, X.; Simons, M. Molecular controls of lymphatic VEGFR3 signaling. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 421–429.
- Yarden, Y.; Escobedo, J.A.; Kuang, W.J.; Yang-Feng, T.L.; Daniel, T.O.; Tremble, P.M.; Chen, E.Y.; Ando, M.E.; Harkins, R.N.; Francke, U.; et al. Structure of the receptor for platelet-derived growth factor helps define a family of closely related growth factor receptors. Nature 1986, 323, 226–232.
- Heldin, C.H.; Lennartsson, J.; Westermark, B. Involvement of platelet-derived growth factor ligands and receptors in tumorigenesis. J. Intern. Med. 2018, 283, 16–44.
- Jitariu, A.; Raica, M.; Cîmpean, A.M.; Suciu, S.C. The role of PDGF-B/PDGFR-BETA axis in the normal development and carcinogenesis of the breast. Crit. Rev. Oncol. Hematol. 2018, 131, 46–52.
- Xu, J.; Xie, L.; Guo, W. PDGF/PDGFR effects in osteosarcoma and the “add-on” strategy. Clin. Sarcoma Res. 2018, 8, 15.
- Qian, H.; Appiah-Kubi, K.; Wang, Y.; Wu, M.; Tao, Y.; Wu, Y.; Chen, Y. The clinical significance of platelet-derived growth factors (PDGFs) and their receptors (PDGFRs) in gastric cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2018, 127, 15–28.
- Roskoski, R., Jr. The role of small molecule platelet-derived growth factor receptor (PDGFR) inhibitors in the treatment of neoplastic disorders. Pharmacol. Res. 2018, 129, 65–83.
- Pal, I.; Mandal, M. PI3K and Akt as molecular targets for cancer therapy: Current clinical outcomes. Acta Pharmacol. Sin. 2012, 33, 1441–1458.
- Jang, H.J.; Suh, P.G.; Lee, Y.J.; Shin, K.J.; Cocco, L.; Chae, Y.C. PLCγ1: Potential arbitrator of cancer progression. Adv. Biol. Regul. 2018, 67, 179–189.
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803.
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316.
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934.
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande-Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804.
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande-Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925.
- Sattler, M.; Salgia, R. c-Met and hepatocyte growth factor: Potential as novel targets in cancer therapy. Curr. Oncol. Rep. 2007, 9, 102–108.
- Mo, H.N.; Liu, P. Targeting MET in cancer therapy. Chronic Dis. Transl. Med. 2017, 3, 148–153.
- Ma, P.C.; Maulik, G.; Christensen, J.; Salgia, R. c-Met: Structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003, 22, 309–325.
- Czyz, M. HGF/c-MET Signaling in Melanocytes and Melanoma. Int. J. Mol. Sci. 2018, 19, 3844.
- Noriega-Guerra, H.; Freitas, V.M. Extracellular Matrix Influencing HGF/c-MET Signaling Pathway: Impact on Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3300.
- Zhang, J.; Jiang, X.; Jiang, Y.; Guo, M.; Zhang, S.; Li, J.; He, J.; Liu, J.; Wang, J.; Ouyang, L. Recent advances in the development of dual VEGFR and c-Met small molecule inhibitors as anticancer drugs. Eur. J. Med. Chem. 2016, 108, 495–504.
- Miranda, O.; Farooqui, M.; Siegfried, J. Status of Agents Targeting the HGF/c-Met Axis in Lung Cancer. Cancers (Basel) 2018, 10, 280.
- Abounader, R.; Reznik, T.; Colantuoni, C.; Martinez-Murillo, F.; Rosen, E.M.; Laterra, J. Regulation of c-Met-dependent gene expression by PTEN. Oncogene 2004, 23, 9173–9182.
- Vouri, M.; Hafizi, S. TAM receptor tyrosine kinases in cancer drug resistance. Cancer Res. 2017, 77, 2775–2778.
- Graham, D.; DeRyckere, D.; Davies, K.; Earp, H. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785.
- Caberoy, N.; Alvarado, G.; Bigcas, J.; Li, W. Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 2012, 227, 401–407.
- Cristina, C.; Laura, G.; Cinzia, L.; Nadia, Z.; Diego, C.; Paola, P. Role of the receptor tyrosine kinase Axl and its targeting in cancer cells. Curr. Med. Chem. 2016, 23, 1496–1512.
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910.
- Shen, Y.; Chen, X.; He, J.; Liao, D.; Zu, X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 2018, 198, 99–111.
- Antony, J.; Huang, R.Y. AXL-Driven EMT State as a Targetable Conduit in Cancer. Cancer Res. 2017, 77, 3725–3732.
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83.
- Schoumacher, M.; Burbridge, M. Key Roles of AXL and MER Receptor Tyrosine Kinases in Resistance to Multiple Anticancer Therapies. Curr. Oncol. Rep. 2017, 19, 19.
- Scaltriti, M.; Elkabets, M.; Baselga, J. Molecular Pathways: AXL, a Membrane Receptor Mediator of Resistance to Therapy. Clin. Cancer Res. 2016, 22, 1313–1317.
- Goff, D.; Zhang, J.; Heckrodt, T.; Yu, J.; Ding, P.; Singh, R.; Holland, S.; Li, W.; Irving, M. Discovery of dual Axl/VEGF-R2 inhibitors as potential anti-angiogenic and anti-metastatic drugs for cancer chemotherapy. Bioorg. Med. Chem. Lett. 2017, 27, 3766–3771.
- Plotnikov, A.N.; Hubbard, S.R.; Schlessinger, J.; Mohammadi, M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 2000, 101, 413–424.
- Gong, S.G. Isoforms of receptors of fibroblast growth factors. J. Cell. Physiol. 2014, 229, 1887–1895.
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266.
- Ghedini, G.C.; Ronca, R.; Presta, M.; Giacomini, A. Future applications of FGF/FGFR inhibitors in cancer. Expert Rev. Anticancer Ther. 2018, 18, 861–872.
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267.
- Du, X.; Xie, Y.; Xian, C.J.; Chen, L. Role of FGFs/FGFRs in skeletal development and bone regeneration. J. Cell. Physiol. 2012, 227, 3731–3743.
- Turner, N.; Grose, R. Fibroblast Growth Factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129.
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4853 Tumors by Next-Generation Sequencing. Clin. Cancer. Res. 2016, 22, 259–267.
- Powers, C.J.; McLeskey, S.W.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 2000, 7, 165–197.
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: Biologic and clinical implications. Cancer Metastasis Rev. 2015, 34, 479–496.
- Peng, W.C.; Lin, X.; Torres, J. The strong dimerization of the transmembrane domain of Williams SV the fibroblast growth factor receptor (FGFR) is modulated by C-terminal juxtamembrane residues. Protein Sci. 2009, 18, 450–459.
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813.
- Tannir, N.M.; Arén-Frontera, O.; Hammers, H.J.; Carducci, M.A.; McDermott, D.F.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; Porta, C. Thirty-month follow-up of the phase III CheckMate 214 trial of first-line nivolumab + ipilimumab (N+I) or sunitinib (S) in patients (pts) with advanced renal cell carcinoma (aRCC). J. Clin. Oncol. 2019, 37 (Suppl. 7), 547.
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127.
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
08 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No