 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Judit Jimenez Sainz | + 2989 word(s) | 2989 | 2021-06-02 11:09:08 | | | |
| 2 | Camila Xu | Meta information modification | 2989 | 2021-06-07 05:42:34 | | |
Video Upload Options
Breast Cancer Susceptibility Gene 2 (BRCA2) is an essential protein that mediates the loading of RAD51 onto resected DNA breaks, a key step in HDR.
1. Introduction
Breast Cancer Susceptibility Gene 2 (BRCA2) was identified in 1994 at the Institute of Cancer Research by the Stratton Laboratory [1][2], only one year after the gene mapping of BRCA1 [3]. Germline mutations in the BRCA genes account for 20–60% of breast cancer cases in families with multiple cancer incidence [4]. The lifetime risk for breast and ovarian cancer rises dramatically for BRCA mutation germline carriers compared to the general population (Figure 1). The overall risk varies by family history, type and location of the mutation, age at diagnosis, parity (number of term pregnancies), environmental factors, and genetic modifiers [5][6][7][8][9][10]. The discovery of the hereditary breast and ovarian cancer (HBOC) BRCA genes in the early 1990s laid the framework for the future of precision medicine as patients became empowered with the ability to make life-changing decisions to manage and mitigate cancer risks [11][12].
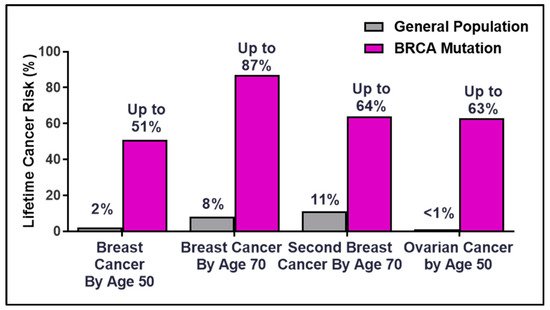
Figure 1. Percentages of lifetime breast and ovarian cancer risks in BRCA germline carriers.
Rapid advances in DNA sequencing technology coupled with public awareness and quick adoption of genetic testing for HBOC genes in genetic counseling clinics have resulted in vast database repositories of BRCA mutations and variants. While the majority of BRCA mutations are associated with breast, ovarian, pancreatic, and prostate cancers, mutations have been documented in other tumor sites such as lung and malignant melanoma.
Soon after the BRCA gene sequences were determined, Myriad Genetics initiated a vigorous intellectual property protection strategy providing them with the exclusive rights to provide all diagnostic testing. A supreme court decision in 2013 overturned Myriads’ patents and opened up the genetic testing landscape to several commercial and academic laboratories. As a result, thousands of BRCA variants have been deposited into various databases. While the majority of BRCA sequencing information has been released, Myriad still maintains a large database of BRCA variants not accessible by the public research community (https://www.wsj.com/articles/geneticists-call-on-myriad-to-share-proprietary-data-to-aid-gene-tests-11578851248, accessed on 5 May 2021).
Genetic testing of patient germline DNA and primary tumor DNA (or circulating tumor DNA (ctDNA)) has resulted in more than 30,000 genetic changes in the BRCA genes that lack functional interpretation. This knowledge gap has led to uncertainty in the ability of genetic counselors to predict cancer risk and impedes the successful clinical management of patients who may benefit from targeted therapies. International collaborations and consortiums have joined forces to design integrative approaches to combine familial and clinical data, in silico predictions, and functional analysis of BRCA2 variants of uncertain significance (VUS). However, the challenge to evaluate extremely rare VUS looms large and functional assays may be the only avenue for some patients to rely on for fundamental life-changing preventative clinical options.
2. BRCA2 Protein, Functions, and Patient Mutations
BRCA2 plays a key role in DNA damage repair via HDR, a high-fidelity repair pathway for DNA DSB repair [13][14]. In mammalian cells, DNA DSBs can be repaired by two major pathways, non-homologous end joining or HDR. HDR requires a template, usually the sister chromatid, and is thus restricted to the S or G2 phase of the cell cycle. If a cell encounters a DNA DSB and commits to the HDR pathway, several nucleases will resect the DNA end resulting in single-stranded DNA (ssDNA) 3’ terminated tails. The ssDNA is immediately coated with Replication Protein A (RPA) to remove the secondary structure and prevent self-annealing. BRCA2 then delivers RAD51 to the ssDNA displacing the RPA protein while simultaneously preventing RAD51 from binding to dsDNA and downregulating the ATPase activity of RAD51 [15]. These mediator activities of BRCA2 result in nucleation and stabilization of RAD51 nucleoprotein filaments on ssDNA, a necessary step preceding RAD51-dependent DNA strand invasion into a homologous donor [15][16][17][18][19][20][21]. Another interesting feature of BRCA2 is its role in replication fork protection. Utilizing the DNA fiber combing technique, several groups have found that under conditions of replicative stress, BRCA2 deficient cells undergo degradation of nascent DNA [22][23][24][25][26]. The hypothesis is that RAD51 is unable to protect the annealed nascent DNA strands at a reversed fork in the absence of BRCA2, and unchecked nucleolytic degradation by MRE11 and other resection nucleases leads to fork collapse and DNA DSBs [25]. A separation-of-function mutation located in the C-terminal domain of BRCA2 (Figure 2), S3291A, was shown to have a defect in fork protection while remaining functionally competent for HDR [25]. Described in detail elsewhere, an ever-growing list of functions has been ascribed to BRCA2, such as R loop processing, gap suppression following DNA replication, and cytokinesis [27][28][29][30][31].
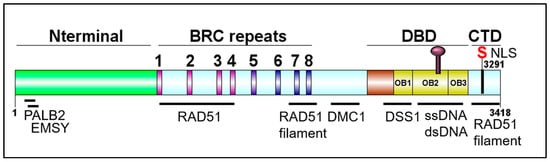
Figure 2. Schematic of BRCA2 domains. DBD: DNA binding domain; CTD: C-terminal domain. DOG illustrator of protein domain structure [32].
The BRCA2 genomic locus is 84 kb, contains 27 exons, and is located on chromosome 13 [12]. The BRCA2 gene encodes a 3418-amino acid nuclear protein with four distinct domains: An N-terminal domain containing residues important for interactions with PALB2 and EMSY, a central BRC repeats region, a DNA binding domain (DBD) containing three tandem oligonucleotide/oligosaccharide-binding folds (OB-folds), and a C-terminal domain (CTD) (Figure 2). PALB2 (Partner and Localizer of BRCA2) mediates the interaction between BRCA1 and BRCA2, plays a role in the nuclear localization of BRCA2, aids in RAD51 loading at resected DNA breaks, and pathogenic mutations in PALB2 have been found to predispose to breast, ovarian, and prostate cancer. EMSY is a BRCA2-interacting transcriptional repressor that intriguingly is linked to sporadic breast and ovarian cancer [33][34][35][36][37]. The eight BRC repeats play critical roles in binding and regulating RAD51 loading and filament stability [38][39][40]. The CTD also functions in binding and stabilizing the RAD51 nucleoprotein filament. BRCA2 plays an important role in meiosis, and hence, binds the meiotic counterpart to RAD51, DMC1 [41]. DSS1 is a small acidic protein visible in the crystal structure of the DBD, makes several intimate contacts with BRCA2, and has been found to facilitate RPA displacement [42][43][44]. Together, all of the protein and DNA interaction domains of BRCA2 promote successful HDR and fork protection [33][36][38][39][44][45][46][47][48][49][50].
It is essential that BRCA2 localizes to the nucleus to carry out its genome integrity and tumor suppressor functions, and thus, contains several putative nuclear localization signals (NLS) [51][52][53][54]. Surprisingly, only a handful of studies have characterized both the NLS and nuclear export signals (NES) of BRCA2, and it remains unclear which specific sequences are necessary and sufficient for the proper regulation of BRCA2 nuclear/cytoplasmic trafficking [51][52][53][54][55].
In cellular models, disruption of BRCA2 leads to the accumulation of DNA DSBs, a severe decrease in HDR activity, and consequently, sensitivity to DNA damaging agents that induce replication fork stalling and/or DNA DSBs such as crosslinking agents or PARP inhibitors (PARPi) [45][56][57][58]. In addition, BRCA2 deficiency leads to micronuclei formation and centrosome amplification, ultimately contributing to genomic instability [59]. BRCA2 knockout mice are embryonic lethal, and only a handful of cell lines (VC8 (hamster); DLD1, CAPAN1, PEO1 (human)) are viable without the expression of full-length protein supporting the essential role that BRCA2 plays in early development and viability [60][61][62][63][64].
Cancer-associated mutations in BRCA2 are commonly found in the BRC repeat and OB-fold regions [9][65][66]. Population-based studies identified more than 10 BRCA2 founder mutations including: 6174delT (BRC repeat region) in Ashkenazi Jews and 7252C>T (DBD region) in the Korean and Finnish populations [67][68][69][70]. BRCA2 mutations found to increase cancer risk consist of indels, frameshifts, and nonsense mutations, ultimately leading to loss of expression or a truncated protein product. As the putative NLS signals of BRCA2 are located at the C-terminus of the protein, most truncated protein products will be mislocalized to the cytoplasm rendering the cells functionally null. Importantly, mutations that lead to loss of nuclear localization of BRCA2 are associated with HDR dysfunction and cancer predisposition [53][62].
The molecular route to tumorigenesis in BRCA2 mutant germline carriers remains an unsolved mystery. The majority of BRCA2 deficient tumors have lost the wild-type allele in a classic case of loss-of-heterozygosity (LOH), however, exceptions to the Knudson’s two-hit hypothesis rule have been described [71]. A generally accepted belief is that BRCA2 haploinsufficiency provides a chronic state of low-level genomic instability conducive to the accumulation of mutations that eventually drive the tumorigenic process. As BRCA2 functions seem necessary for cellular viability, the specific genetic background that is permissive for tumor growth is unknown. Furthermore, the genetic and pathological basis for why BRCA2 (and BRCA1) mutations lead preferentially to cancer of the breast and ovaries remains an ever-present enigma.
The ClinVar database contains over 13,000 reports of germline and somatic BRCA2 alterations with more than 5000 VUS (Figure 3 left panel). Approximately 6000 of the 9500 BRCA2 molecular mutations reported are single amino acid changes in the 3,418-amino acid BRCA2 protein (Figure 3 right panel). Many of the mutations evaluated and reviewed as pathogenic are indeed frameshift and nonsense mutations resulting in either a truncated protein or complete lack of BRCA2 expression.
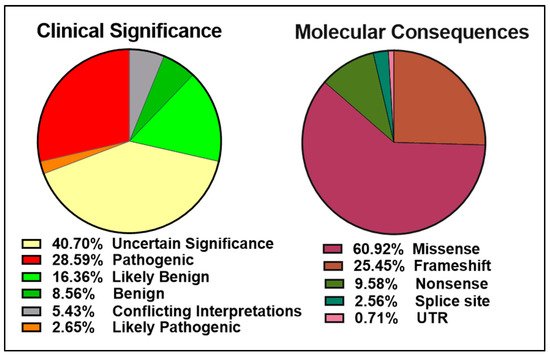
Figure 3. Pie charts representing the percentages of BRCA2 germline and somatic variants grouped into clinical significance (left) or molecular consequences (right) as reported in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/?term=BRCA2%5Bgene%5D, accessed on 10 April 2021).
The tumor spectrum of cancers associated with pathogenic mutations in BRCA2 include: breast, ovarian, fallopian tube, melanoma, prostate, pancreatic, and lung cancers (Figure 4A). The display of all BRCA2 germline and somatic mutations described in the curated set of non-redundant studies of cBioPortal for Cancer Genomics shows that BRCA2 is altered in 10–15% of breast, ovarian, and prostate cancer cases, whereas BRCA2 is altered in 18% of melanoma cases (Figure 4A). The location of BRCA2 mutations in the dataset does not suggest specific “hot-spots” or “cold-spots” but widespread alterations across the whole gene (Figure 4B). As of 2020, a new database, HotSpotsAnnotations, applied a statistical model to detect putative hotspots on The Cancer Genome Atlas (TCGA) cancer datasets which contain 10,182 patients and 33 cancer types. In the cancer HotSpots Database, six BRCA2 mutations were identified at positions 1393, 1689, 1782, 2842, 3308, and 3342 (http://bioinformatica.mty.itesm.mx:8080/HotSpotsAnnotations/PiquinSpot.jsp?gene=BRCA2, accessed on 25 April 2021) [72]. The first three were located in the BRC domain, while the latter were mapped to the DNA binding domain. Controversy remains as some studies demonstrated no correlation, and others, a positive correlation between BRCA2 mutations in the BRC domain and ovarian cancer patient survival [73][74]. Discrepancies arise in part due to the selection of the BRCA2 mutations (missense, indels, etc.), the number of patients included, or the treatment provided. A single study in 2020 classified BRCA2 exon 10 and 11 as “cold-spots”, or more tolerant to variation [75]. However, the analysis was based on limited data, and thus overall, additional studies are needed regarding the frequency of mutation locations across the BRCA2 genomic locus.
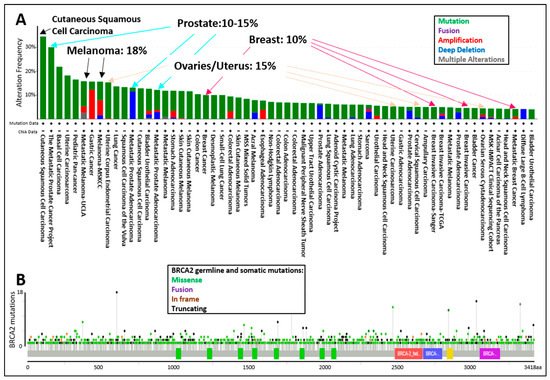
Figure 4. cBioPortal for Cancer Genomics display of the (A) percentage of BRCA2 germline and somatic alterations and (B) spectrum of BRCA2 germline and somatic mutations in the different protein domains. The curated set of non-redundant studies of cBioPortal for Cancer Genomics was used. Combined study with 48,081 samples and 184 studies. Graphs display BRCA2 somatic mutation frequency of 3% and BRCA2 germline mutation frequency of <0.1.
The data obtained from cBioPortal indicate a total of 1934 BRCA2 germline and somatic alterations in the curated set of non-redundant studies. Of the 518 driver mutations, 83.6% are truncations (Figure 5 left panel), whereas missense mutations make up 96.7% (1369) of VUS (1416) (Figure 5 right panel). While truncating mutations in BRCA2 are clearly pathogenic in the majority of cases, the curated data highlight the need to further understand missense mutations, as they represent the most abundant VUS.
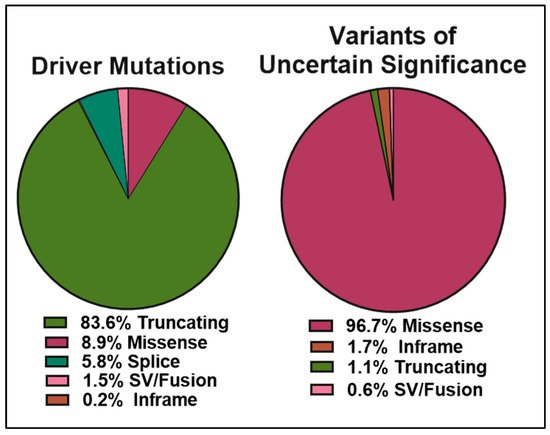
Figure 5. Pie charts representing the percentages of BRCA2 germline and somatic genetic variants grouped into driver mutations (left) and VUS (right) as reported in cBioPortal for Cancer Genomics database (https://www.cbioportal.org/results/cancerTypesSummary?tab_index=tab_visualize&Action=Submit&session_id=607f022de4b0242bd5d49461, accessed on 10 April 2021). The curated set of non-redundant studies of cBioPortal for Cancer Genomics was used. A total of 48,081 samples and 184 studies. The total number of driver mutations and variants of uncertain significance (1934) corresponds to 3% of the samples with BRCA2 somatic mutations and <0.1% of the samples with BRCA2 germline mutations.
3. Clinical Management of Patients
The American College of Obstetrician and Gynecologists (ACOG) and the American Cancer Society have created guidelines for clinicians to manage patients with BRCA2 gene mutations (Figure 6) [76][77].
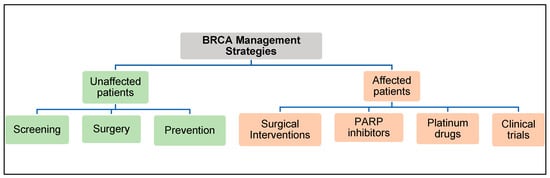
Figure 6. BRCA2 Management Strategies.
Sequencing-based genetic tests to screen for BRCA mutations are offered to individuals with a family history of breast or ovarian cancer. As a risk-reduction strategy, pathogenic BRCA mutation carriers, who are cancer-free, will be informed of surgical interventions such as a double mastectomy and/or unilateral salpingo-oophorectomy. In many cases, female BRCA carriers during their early reproductive years may opt for careful surveillance in order to delay prophylactic surgeries that would prevent them from having children. In 2013, the so-called “Angelina effect” sparked an acute public awareness of genetic testing for hereditary cancer risk genes, which may have resulted in many people undergoing unnecessary testing without significant associated risk factors such as a family history of unusually high cancer incidence [78][79]. In a worst-case scenario, a recent article in the Wall Street Journal described a family where multiple women underwent prophylactic surgeries based on a “likely” pathogenic BRCA VUS finding that was later re-classified as a benign variant (https://www.wsj.com/articles/seven-women-in-a-family-chose-surgery-after-a-genetic-test-then-the-results-changed-11576860210, accessed on 24 April 2021). This failure of precision medicine underscores the need to cautiously interpret VUS findings and to bolster VUS evaluations with additional evidence for pathogenicity. We suggest that rigorous functional assays performed by laboratories with expertise in BRCA2 biology could provide meaningful criteria for evaluating rare VUS when familial, co-segregation, co-occurrence, and other classical genetic linkage data are lacking.
For patients who learn they are BRCA2 VUS carriers, the situation is complex and frustrating. Not only does the patient now know their BRCA2 gene is not normal (deviates from the wild-type sequence), the clinical significance of the VUS findings are rarely known, leaving the patient with unknown future cancer risk. Universally accepted standards for how to deal with BRCA VUS have yet to be established, revealing a shortcoming in our ability to make informed clinical decisions [78][79][80]. Thus, it is critical that genetic counselors have the ability to intercede and minimally inform patients of their options thus that rational choices can be made.
Patients diagnosed with cancer and for whom a germline or somatic pathogenic BRCA2 mutation is found often respond well to chemotherapy agents such as PARPi and platinum drugs. LOH of the wild-type BRCA2 allele is usually associated with a robust clinical response to these drugs but not in every case [71]. A tumor that is classified as HDR deficient is likely the best biomarker correlating with clinical response to platinum drugs and PARPi. Some patients still fail to respond to these drugs, and others may initially respond, but ultimately relapse as resistant tumor cells emerge and recolonize the tumor. Clinical trials with inhibitors such as AZD2281, 6-Mercaptopurine, and methotrexate are ongoing for BRCA2 defective ovarian tumors focused on resistance. Somatic findings of BRCA2 VUS in a tumor present a similar challenge to germline findings. In a tumor scenario, LOH of the wild-type BRCA2 allele may determine whether a patient is a good candidate for PARPi therapy. Again, if the tumor can be classified as HDR deficient, this result would provide the motivation for the use of synthetic lethal PARPi therapy. In summary, BRCA2 VUS, whether germline or somatic, presents an ongoing problem for clinical decision-making.
4. Curation of BRCA2 VUS
Several international groups and consortiums have joined efforts to create databases reporting BRCA2 genetic variants generated from sequencing analysis (Table 1 and Table 2). While publicly available databases are useful resources, curation of the information and classification of the variants is an ongoing challenge.
Table 1. International groups investigating BRCA mutations in various populations.
| Study/Consortium | Description |
|---|---|
| CIMBA | Consortium of investigators of modifiers of BRCA1/2 was established in 2005 |
| PROSE | Consortium in the prevention and observation of surgical endpoints [81] |
| ENIGMA | International consortium of investigators to determine the clinical significance of sequence variants in BRCA1, BRCA2 and other known or suspected breast cancer genes (www.enigmaconsortium.org, accessed on 15 April 2021) [82] |
| EMBRACE | Study aims to create a register of BRCA1 and BRCA2 families with defects in other genes to find out the cancer risk in these associations [10] |
| IBCCS | International BRCA1/2 carrier cohort study: purpose, rationale, and study design (https://breast-cancer-research.biomedcentral.com/articles/10.1186/bcr93, accessed on 15 April 2021) |
| Breast Cancer Linkage Consortium | International data sharing platform established in 1989 to collect families with breast cancer by linkage analysis |
| PROMPT | Prospective Registry of Multiplex Testing for patients and their families (www.promptstudy.org, accessed on 15 April 2021) |
Table 2. Clinical Significance Databases with BRCA2 mutations (Updated April 2021).
| BRCA2 Databases | Total Variants | Variants of Uncertain Significance | Missense Variants | Pathogenic Variants |
|---|---|---|---|---|
| Breast Cancer Information Core (BIC) (https://research.nhgri.nih.gov/bic/, accessed on 15 April 2021) |
14,914 |  |
7156 (48%) | |
| Leiden Open Variant Database (LOVD) (https://www.lovd.nl, accessed on 15 April 2021) |
920 | |
871 (94.6%) | |
| Catalogue of Somatic Mutation in Cancer (COSMIC) (https://cancer.sanger.ac.uk/cosmic, accessed on 15 April 2021) |
2729 | |
1548 (56.72%) | |
| BRCA Exchange (https://brcaexchange.org, accessed on 15 April 2021) |
20,751 | 16,649 (83%) | |
2672 (13%) |
| ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accessed on 15 April 2021) |
13,109 | 5381 (41%) | 5756 (44%) | 3780 (29%) |
Data unavailable.
There are several commonly used online database resources that provide some interpretation of BRCA2 sequence variants (Table 2). For example, BRCA Exchange was created by the Global Alliance for Genomics and Health (GA4GH) and is the largest open-source database on BRCA variant information [83]. BRCA Exchange contains information from ClinVar, the Breast Cancer Information Core database, Leiden Open Variant Database (LOVD), several population databases, and is conveniently accessible through a smartphone app. Many of these databases, such as ClinVar and LOVD, are freely accessible and contain submissions reporting BRCA variants found in patient samples [84]. These databases display BRCA variants as expert-reviewed, interpreted, and classified by supporting evidence such as familial cases and co-segregation studies. Some of the databases display the BRCA information by molecular alteration and/or clinical significance (Table 2). The comparison of five databases with BRCA2 information shows that missense mutations are around 44–96% of the data deposited in the databases, and BRCA2 VUS make up 41–83% of ClinVar and BRCA Exchange databases. Thus, although collaborative efforts have unified the information of BRCA2 mutations, most remain unknown for cancer risk.
References
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene brca2. Nature 1995, 378, 789–792.
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994, 265, 2088–2090.
- Bowcock, A.; Anderson, L.A.; Friedman, L.S.; Black, D.M.; Osborne-Lawrence, S.; Rowell, S.E.; Hall, J.M.; Solomon, E.; King, M.C. THRA1 and D17S183 flank an interval of <4 cm for the breast-ovarian cancer gene (BRCA1) on chromosome 17q21. Am. J. Hum. Genet. 1993, 52, 718–722.
- Nathanson, K.L.; Wooster, R.; Weber, B.L. Breast cancer genetics: What we know and what we need. Nat. Med. 2001, 7, 552–556.
- Andrieu, N.; Goldgar, D.E.; Easton, D.F.; Rookus, M.; Brohet, R.; Antoniou, A.C.; Chang-Claude, J. Pregnancies, breast-feeding, and breast cancer risk in the international brca1/2 carrier cohort study (ibccs). J. Natl. Cancer Inst. 2006, 98, 535–544.
- Cullinane, C.A.; Lubinski, J.; Neuhausen, S.L.; Ghadirian, P.; Lynch, H.T.; Isaacs, C.; Weber, B.; Moller, P.; Offit, K.; Kim-Sing, C.; et al. Effect of pregnancy as a risk factor for breast cancer inBRCA1/BRCA2 mutation carriers. Int. J. Cancer 2005, 117, 988–991.
- Metcalfe, K.; Lubinski, J.; Lynch, H.T.; Ghadirian, P.; Foulkes, W.D.; Kim-Sing, C.; Neuhausen, S.; Tung, N.; Rosen, B.; Gronwald, J.; et al. Family History of Cancer and Cancer Risks in Women with BRCA1 or BRCA2 Mutations. J. Natl. Cancer Inst. 2010, 102, 1874–1878.
- Milne, R.L.; Osorio, A.; Cajal, T.R.Y.; Baiget, M.; Lasa, A.; Diaz-Rubio, E.; De La Hoya, M.; Caldés, T.; Teulé, A.; Lázaro, C.; et al. Parity and the risk of breast and ovarian cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res. Treat. 2009, 119, 221–232.
- Thompson, D.; Easton, D. Variation in Cancer Risks, by Mutation Position, in BRCA2 Mutation Carriers. Am. J. Hum. Genet. 2001, 68, 410–419.
- Mavaddat, N.; Peock, S.; Frost, D.; Ellis, S.; Platte, R.; Fineberg, E.; Evans, D.G.; Izatt, L.; Eeles, R.A.; Adlard, J.; et al. Cancer Risks for BRCA1 and BRCA2 Mutation Carriers: Results From Prospective Analysis of EMBRACE. J. Natl. Cancer Inst. 2013, 105, 812–822.
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.; Mooij, T.M.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; Goldgar, D.E.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416.
- Easton, D.F.; Steele, L.; Fields, P.; Ormiston, W.; Averill, D.; Daly, P.A.; McManus, R.; Neuhausen, S.L.; Ford, D.; Wooster, R.; et al. Cancer Risks in Two Large Breast Cancer Families Linked to BRCA2 on Chromosome 13q12-13. Am. J. Hum. Genet. 1997, 61, 120–128.
- Scully, R.; Livingston, D.M. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nat. Cell Biol. 2000, 408, 429–432.
- Venkitaraman, A.R. Cancer Susceptibility and the Functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182.
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683.
- Davies, A.A.; Masson, J.-Y.; McIlwraith, M.J.; Stasiak, A.Z.; Stasiak, A.; Venkitaraman, A.R.; West, S.C. Role of BRCA2 in Control of the RAD51 Recombination and DNA Repair Protein. Mol. Cell 2001, 7, 273–282.
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.-D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262.
- Powell, S.N.; Willers, H.; Xia, F. Brca2 keeps rad51 in line. High-fidelity homologous recombination prevents breast and ovarian cancer? Mol. Cell 2002, 10, 1262–1263.
- Thorslund, T.; McIlwraith, M.J.; Compton, S.A.; Lekomtsev, S.; Petronczki, M.; Griffith, J.D.; West, S.C. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1263–1265.
- Yang, H.; Li, Q.; Fan, J.; Holloman, W.K.; Pavletich, N.P. The brca2 homologue brh2 nucleates rad51 filament formation at a dsdna-ssdna junction. Nature 2005, 433, 653–657.
- Yuan, S.S.; Lee, S.Y.; Chen, G.; Song, M.; Tomlinson, G.E.; Lee, E.Y. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999, 59, 3547–3551.
- Eckelmann, B.J.; Bacolla, A.; Wang, H.; Ye, Z.; Guerrero, E.N.; Jiang, W.; El-Zein, R.; Hegde, M.L.; Tomkinson, A.E.; Tainer, J.A.; et al. XRCC1 promotes replication restart, nascent fork degradation and mutagenic DNA repair in BRCA2-deficient cells. NAR Cancer 2020, 2, zcaa013.
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Lopes, M. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859.
- Rickman, K.A.; Noonan, R.J.; Lach, F.; Sridhar, S.; Wang, A.; Abhyankar, A.; Huang, A.; Kelly, M.; Auerbach, A.D.; Smogorzewska, A. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes Dev. 2020, 34, 832–846.
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542.
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.-W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nat. Cell Biol. 2014, 511, 362–365.
- Shivji, M.K.; Renaudin, X.; Williams, Ç.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039.
- Daniels, M.J.; Wang, Y.; Lee, M.; Venkitaraman, A.R. Abnormal Cytokinesis in Cells Deficient in the Breast Cancer Susceptibility Protein BRCA2. Science 2004, 306, 876–879.
- Mondal, G.; Rowley, M.; Guidugli, L.; Wu, J.; Pankratz, V.S.; Couch, F.J. BRCA2 Localization to the Midbody by Filamin A Regulates CEP55 Signaling and Completion of Cytokinesis. Dev. Cell 2012, 23, 137–152.
- Panzarino, N.J.; Krais, J.J.; Cong, K.; Peng, M.; Mosqueda, M.; Nayak, S.U.; Bond, S.M.; Calvo, J.A.; Doshi, M.B.; Bere, M.; et al. Replication Gaps Underlie BRCA Deficiency and Therapy Response. Cancer Res. 2021, 81, 1388–1397.
- Ren, J.; Wen, L.; Gao, X.; Jin, C.; Xue, Y.; Yao, X. DOG 1.0: Illustrator of protein domain structures. Cell Res. 2009, 19, 271–273.
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol. Cell 2006, 22, 719–729.
- Dray, E.; Etchin, J.; Wiese, C.; Saro, D.; Williams, G.J.; Hammel, M.; Yu, X.; Galkin, V.E.; Liu, D.; Tsai, M.-S.; et al. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat. Struct. Mol. Biol. 2010, 17, 1255–1259.
- Foo, T.K.; Tischkowitz, M.; Simhadri, S.; Boshari, T.; Zayed, N.; Burke, K.A.; Berman, S.H.; Blecua, P.; Riaz, N.; Huo, Y.; et al. Compromised BRCA1–PALB2 interaction is associated with breast cancer risk. Oncogene 2017, 36, 4161–4170.
- Hughes-Davies, L.; Huntsman, D.; Ruas, M.; Fuks, F.; Bye, J.; Chin, S.-F.; Milner, J.; Brown, L.; Hsu, F.; Gilks, B.; et al. EMSY Links the BRCA2 Pathway to Sporadic Breast and Ovarian Cancer. Cell 2003, 115, 523–535.
- Tischkowitz, M.; Capanu, M.; Sabbaghian, N.; Li, L.; Liang, X.; Vallée, M.P.; Tavtigian, S.V.; Concannon, P.; Foulkes, W.D.; Bernstein, L.; et al. Rare germline mutations inPALB2and breast cancer risk: A population-based study. Hum. Mutat. 2012, 33, 674–680.
- Carreira, A.; Hilario, J.; Amitani, I.; Baskin, R.J.; Shivji, M.K.; Venkitaraman, A.R.; Kowalczykowski, S.C. The BRC Repeats of BRCA2 Modulate the DNA-Binding Selectivity of rad51. Cell 2009, 136, 1032–1043.
- Chatterjee, G.; Jimenez-Sainz, J.; Presti, T.; Nguyen, T.; Jensen, R.B. Distinct binding of BRCA2 BRC repeats to RAD51 generates differential DNA damage sensitivity. Nucleic Acids Res. 2016, 44, 5256–5270.
- Carreira, A.; Kowalczykowski, S.C. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 10448–10453.
- Thorslund, T.; Esashi, F.; West, S.C. Interactions between human BRCA2 protein and the meiosis-specific recombinase DMC1. EMBO J. 2007, 26, 2915–2922.
- Marston, N.J.; Richards, W.J.; Hughes, D.; Bertwistle, D.; Marshall, C.J.; Ashworth, A. Interaction between the Product of the Breast Cancer Susceptibility Gene BRCA2 and DSS1, a Protein Functionally Conserved from Yeast to Mammals. Mol. Cell. Biol. 1999, 19, 4633–4642.
- Zhao, W.; Vaithiyalingam, S.; Filippo, J.S.; Maranon, D.G.; Jimenez-Sainz, J.; Fontenay, G.V.; Kwon, Y.; Leung, S.G.; Lu, L.; Jensen, R.B.; et al. Promotion of BRCA2-Dependent Homologous Recombination by DSS1 via RPA Targeting and DNA Mimicry. Mol. Cell 2015, 59, 176–187.
- Yang, H.; Jeffrey, P.D.; Miller, J.; Kinnucan, E.; Sun, Y.; Thomä, N.H.; Zheng, N.; Chen, P.-L.; Lee, W.-H.; Pavletich, N.P. BRCA2 Function in DNA Binding and Recombination from a BRCA2-DSS1-ssDNA Structure. Science 2002, 297, 1837–1848.
- Chen, P.-L.; Chen, C.-F.; Chen, Y.; Xiao, J.; Sharp, Z.D.; Lee, W.-H. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl. Acad. Sci. USA 1998, 95, 5287–5292.
- Mizuta, R.; LaSalle, J.; Cheng, H.-L.; Shinohara, A.; Ogawa, H.; Copeland, N.; Jenkins, N.A.; Lalande, M.; Alt, F.W. RAB22 and RAB163/mouse BRCA2: Proteins that specifically interact with the RAD51 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6927–6932.
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51–BRCA2 complex. Nat. Cell Biol. 2002, 420, 287–293.
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.-S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nat. Cell Biol. 1997, 386, 804–810.
- Wong, A.K.C.; Pero, R.; Ormonde, P.A.; Tavtigian, S.V.; Bartel, P.L. RAD51 Interacts with the Evolutionarily Conserved BRC Motifs in the Human Breast Cancer Susceptibility Gene brca2. J. Biol. Chem. 1997, 272, 31941–31944.
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996.
- Bertwistle, D.; Swift, S.; Marston, N.J.; Jackson, L.E.; Crossland, S.; Crompton, M.R.; Marshall, C.J.; Ashworth, A. Nuclear location and cell cycle regulation of the BRCA2 protein. Cancer Res. 1997, 57, 5485–5488.
- Han, X.; Saito, H.; Miki, Y.; Nakanishi, A. A CRM1-mediated nuclear export signal governs cytoplasmic localization of BRCA2 and is essential for centrosomal localization of BRCA2. Oncogene 2007, 27, 2969–2977.
- Spain, B.H.; Larson, C.J.; Shihabuddin, L.S.; Gage, F.H.; Verma, I.M. Truncated BRCA2 is cytoplasmic: Implications for cancer-linked mutations. Proc. Natl. Acad. Sci. USA 1999, 96, 13920–13925.
- Yano, K.-I.; Morotomi, K.; Saito, H.; Kato, M.; Matsuo, F.; Miki, Y. Nuclear Localization Signals of the BRCA2 Protein. Biochem. Biophys. Res. Commun. 2000, 270, 171–175.
- Jeyasekharan, A.; Liu, Y.; Hattori, H.; Pisupati, V.; Jonsdottir, A.B.; Rajendra, E.; Lee, M.; Sundaramoorthy, E.; Schlachter, S.; Kaminski, C.F.; et al. A cancer-associated BRCA2 mutation reveals masked nuclear export signals controlling localization. Nat. Struct. Mol. Biol. 2013, 20, 1191–1198.
- Chen, J.J.; Silver, D.; Cantor, S.; Livingston, D.M.; Scully, R. BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res. 1999, 59, 1752–1756.
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 Is Required for Homology-Directed Repair of Chromosomal Breaks. Mol. Cell 2001, 7, 263–272.
- Patel, K.J.; Yu, V.P.; Lee, H.; Corcoran, A.; Thistlethwaite, F.C.; Evans, M.J.; Colledge, W.H.; Friedman, L.S.; Ponder, B.A.; Venkitaraman, A.R. Involvement of Brca2 in DNA Repair. Mol. Cell 1998, 1, 347–357.
- Tutt, A.; Gabriel, A.; Bertwistle, D.; Connor, F.; Paterson, H.; Peacock, J.; Ross, G.; Ashworth, A. Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr. Biol. 1999, 9, 1107–1110.
- Chen, J.; Silver, D.P.; Walpita, D.; Cantor, S.B.; Gazdar, A.F.; Tomlinson, G.; Couch, F.J.; Weber, B.L.; Ashley, T.; Livingston, D.M.; et al. Stable Interaction between the Products of the BRCA1 and BRCA2 Tumor Suppressor Genes in Mitotic and Meiotic Cells. Mol. Cell 1998, 2, 317–328.
- Hucl, T.; Rago, C.; Gallmeier, E.; Brody, J.R.; Gorospe, M.; Kern, S.E. A Syngeneic Variance Library for Functional Annotation of Human Variation: Application to BRCA2. Cancer Res. 2008, 68, 5023–5030.
- Sakai, W.; Swisher, E.M.; Jacquemont, C.; Chandramohan, K.V.; Couch, F.J.; Langdon, S.P.; Wurz, K.; Higgins, J.; Villegas, E.; Taniguchi, T. Functional Restoration of BRCA2 Protein by Secondary BRCA2 Mutations in BRCA2-Mutated Ovarian Carcinoma. Cancer Res. 2009, 69, 6381–6386.
- Suzuki, A.; De La Pompa, J.L.; Hakem, R.; Elia, A.; Yoshida, R.; Mo, R.; Nishina, H.; Chuang, T.; Wakeham, A.; Itie, A.; et al. Brca2 is required for embryonic cellular proliferation in the mouse. Genes Dev. 1997, 11, 1242–1252.
- Wiegant, W.W.; Overmeer, R.M.; Godthelp, B.C.; van Buul, P.P.; Zdzienicka, M.Z. Chinese hamster cell mutant, V-C8, a model for analysis of Brca2 function. Mutat. Res. Mol. Mech. Mutagen. 2006, 600, 79–88.
- Guidugli, L.; Carreira, A.; Caputo, S.M.; Ehlen, A.; Galli, A.; Monteiro, A.N.; Neuhausen, S.L.; Hansen, T.V.; Couch, F.J.; Vreeswijk, M.P.; et al. Functional Assays for Analysis of Variants of Uncertain Significance inBRCA2. Hum. Mutat. 2014, 35, 151–164.
- Karchin, R.; Agarwal, M.; Sali, A.; Couch, F.; Beattie, M.S. Classifying Variants of Undetermined Significance in BRCA2 with Protein Likelihood Ratios. Cancer Inform. 2008, 6, 203–216.
- Abul-Husn, N.S.; Team, C.G.; Soper, E.; Odgis, J.A.; Cullina, S.; Bobo, D.; Moscati, A.; Rodriguez, J.E.; Loos, R.; Cho, J.H.; et al. Exome sequencing reveals a high prevalence of BRCA1 and BRCA2 founder variants in a diverse population-based biobank. Genome Med. 2019, 12, 2.
- Neuhausen, S.L.; Gilewski, T.; Norton, L.; Tran, T.; McGuire, P.; Swensen, J.; Hampel, H.; Borgen, P.I.; Brown, K.L.; Skolnick, M.; et al. Recurrent BRCA2 6174delT mutations in Ashkenazi Jewish women affected by breast cancer. Nat. Genet. 1996, 13, 126–128.
- Sarantaus, L.; Huusko, P.; Eerola, H.; Launonen, V.; Vehmanen, P.; Rapakko, K.; Gillanders, E.M.; Syrjäkoski, K.; Kainu, T.; Vahteristo, P.; et al. Multiple founder effects and geographical clustering of BRCA1 and BRCA2 families in Finland. Eur. J. Hum. Genet. 2000, 8, 757–763.
- Seong, M.-W.; Cho, S.; Noh, D.-Y.; Han, W.; Kim, S.-W.; Park, C.-M.; Park, H.-W.; Kim, J.Y.; Park, S.S. Comprehensive mutational analysis ofBRCA1/BRCA2for Korean breast cancer patients: Evidence of a founder mutation. Clin. Genet. 2009, 76, 152–160.
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 2017, 8, 319.
- Trevino, V. Hotspotannotations-a database for hotspot mutations and annotations in cancer. Database (Oxford) 2020, 2020, baaa025.
- Alsop, K.; Fereday, S.; Meldrum, C.; DeFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation–Positive Women With Ovarian Cancer: A Report From the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663.
- Labidi-Galy, S.I.; Olivier, T.; Rodrigues, M.; Ferraioli, D.; Derbel, O.; Bodmer, A.; Petignat, P.; Rak, B.; Chopin, N.; Tredan, O.; et al. Location of Mutation in BRCA2 Gene and Survival in Patients with Ovarian Cancer. Clin. Cancer Res. 2018, 24, 326–333.
- Dines, J.N.; Shirts, B.H.; Slavin, T.P.; Walsh, T.; King, M.-C.; Fowler, D.M.; Pritchard, C.C.; Shirts, B.H.; Pritchard, C.C. Systematic misclassification of missense variants in BRCA1 and BRCA2 “coldspots”. Genet. Med. 2020, 22, 825–830.
- Pruthi, S.; Gostout, B.S.; Lindor, N.M. Identification and management of women with brca mutations or hereditary predisposition for breast and ovarian cancer. Mayo Clin. Proc. 2010, 85, 1111–1120.
- Salhab, M.; Bismohun, S.; Mokbel, K. Risk-reducing strategies for women carrying brca1/2 mutations with a focus on prophylactic surgery. BMC Womens Health 2010, 10, 28.
- Evans, D.G.R.; Barwell, J.; Eccles, D.M.; Collins, A.; Izatt, L.; Jacobs, C.; Donaldson, A.; Brady, A.F.; Cuthbert, A.; Harrison, R.; et al. The Angelina Jolie effect: How high celebrity profile can have a major impact on provision of cancer related services. Breast Cancer Res. 2014, 16, 442.
- James, P.A.; Mitchell, G.; Bogwitz, M.; Lindeman, G.J. The Angelina Jolie effect. Med. J. Aust. 2013, 199, 646.
- Welsh, J.L.; Hoskin, T.L.; Day, C.N.; Thomas, A.S.; Cogswell, J.A.; Couch, F.J.; Boughey, J.C. Clinical Decision-Making in Patients with Variant of Uncertain Significance in BRCA1 or BRCA2 Genes. Ann. Surg. Oncol. 2017, 24, 3067–3072.
- Domchek, S.M.; Friebel, T.M.; Singer, C.F.; Evans, D.G.; Lynch, H.T.; Isaacs, C.; Garber, J.E.; Neuhausen, S.L.; Matloff, E.; Eeles, R.; et al. Association of risk-reducing surgery in brca1 or brca2 mutation carriers with cancer risk and mortality. JAMA 2010, 304, 967–975.
- Spurdle, A.B.; Healey, S.; Devereau, A.; Hogervorst, F.B.L.; Monteiro, A.N.A.; Nathanson, K.L.; Radice, P.; Stoppa-Lyonnet, D.; Tavtigian, S.; Wappenschmidt, B.; et al. ENIGMA-Evidence-based network for the interpretation of germline mutant alleles: An international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 2012, 33, 2–7.
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752.
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844.


