 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tânia S. Morais | + 3346 word(s) | 3346 | 2021-06-03 10:36:37 | | | |
| 2 | Camila Xu | Meta information modification | 3346 | 2021-06-07 05:37:24 | | |
Video Upload Options
Cell adhesion molecules (CAM) are cell surface glycoproteins involved in cell-to-cell and cell-to-extracellular matrix adhesion, a process that is essential for the correct maintenance and function of tissues and organs.
1. Cell Adhesion Molecules (CAM)
Cell adhesion molecules (CAM) are cell surface glycoproteins involved in cell-to-cell and cell-to-extracellular matrix adhesion, a process that is essential for the correct maintenance and function of tissues and organs [1]. CAM are grouped into four different classes—integrins, cadherins, selectins, and the immunoglobulins superfamily. While integrins typically bind to the extracellular matrix, the other three types of CAM are usually associated with cell-to-cell adhesion phenomena [1]. Additionally to their structural function, CAM also act as receptors of a variety of endogenous ligands and messengers, modulating and actively participating in different key biological processes, including cell proliferation and migration, phagocytosis, apoptosis, angiogenesis, and thrombosis [1][2]. Alterations of CAM function, structure, and/or expression patterns are often associated with auto-immune diseases, metabolic syndromes, and cancer [1]. Therefore, CAM have been intensively exploited as potential drug targets, and for instance, some CAM-targeting drugs were already approved for the treatment of patients with thrombosis. Currently, there are also several drug candidates under clinical trials for the treatment of cancer and other disorders [1][2]. Moreover, CAM have also been exploited as targets for targeted drug delivery in precision medicine, given the overexpression of specific CAM in certain diseases, such as cancer, comparatively to healthy tissues. This approach relies on the use of ligands/targeting units (e.g., antibodies, peptides, peptidomimetics, and small molecules, among others) that can recognize and bind with high affinity to the specific type of CAM that is overexpressed in the surface of the tumoral cells as drug carriers, thus being able to selectively delivery the drug into its target while sparing the surrounding tissues [2]. Within this frame, substantial work has been continuously reported, especially regarding the use of integrin- and cadherin-targeting peptides [1].
2. Integrins
Integrins are heterodimeric transmembrane receptors composed of an α- and a β-subunit non-covalently associated with each other, that are dependent of divalent cations such as Mg2+ or Ca2+ for the interaction with their ligands [3][4]. There are 24 subtypes of integrins in mammals, resulting from a limited number of combinations between 18 different α-subunits and 8 diverse β-domains [3]. Additionally to these structural differences, each subtype also has its own cellular distribution, endogenous ligands (e.g., collagen, fibronectin, nephronectin, laminin, etc.), and function [4]. In general, integrins play an important role in a plethora of biological processes by acting as adhesion molecules, mechanosensors, and signal transduction platforms. They can signal both from the extracellular environment into the intracellular compartment as well as in the opposite sense, regulating cell adhesion, proliferation, migration, and survival [3][4]. Integrins also mediate several cancer-related events, such as tumor initiation and progression, malignant transformation, tumor-induced angiogenesis, cancer metastasis and reactivation, and resistance to anticancer immunotherapy [5]. Given the correlation of integrins with the etiology and pathology of several diseases, various integrin-targeting drugs have successfully achieved clinical use and many others are under clinical development, most of them aiming to treat cardiovascular diseases, auto-immune syndromes, and cancer [6]. In the latter, integrins such as αVβ3 and αVβ5 are upregulated in certain types of tumors relative to the other non-tumoral cells, displaying a characteristic distribution in cancer tissues, and/or structural alterations during tumor growth and metastasis [5][7]. Thus, integrins have also been exploited for the targeted drug delivery of anticancer agents by using drug carriers that can selectively bind to these receptors and trigger an integrin-mediated endocytosis process with subsequent accumulation of the drug specifically in the tumor cells [8]. The arginine-glycine-aspartic acid (RGD) motif was found to be present in several natural ligands of the αV-integrin subfamily (such as fibronectin) and to be the minimal sequence needed for appropriate integrin recognition [8][9]. Thus, peptides containing the RGD motif have become a popular tool to the selective delivery of known drugs or drug candidates, including organic small molecules and metal complexes, into integrin-expressing cancer cells for precision therapy and diagnostics [10][11]. Several linear and cyclic peptides have been custom designed as highly specific binders of αVβ3, αVβ6, or α5β1 integrins and many studies regarding their use as delivering vectors for anticancer applications have been reported with promising results [8][9]. However, only few studies of ruthenium and gold complexes vectorized with integrin-targeting agents have been reported. Most of them address the use of the cyclic peptide cyclo-RGDfK (f = D-phenylalanine) that is known to bind selectively and with high affinity to the αVβ3 integrin, but the linear tripeptide RGD (specific for αVβ3/αVβ5) and other RGD-containing sequences were exploited as well. Marchán and co-workers reported the conjugation of the complex [Ru(η6-p-cym)(bpm)(pyac)]2+ (where p-Cym = para-cymene; bpm = 2,2′-bipyrimidine; and pyac = 4-pyridineacetic acid) with the tripeptide RGD using a polyethylene glycol spacer between both moieties (1, Figure 3) [12]. The spacer PEG(2) was selected to improve the aqueous solubility of the conjugate and to keep the ruthenium complex spatially apart from the targeting-peptide so that the activity and the selectivity of each would not be perturbed. Conjugate 1 acts as a prodrug that is stable in aqueous solution at dark, but upon visible light irradiation suffers selective photodissociation from the pyridyl-RGD functionalized ligand, releasing the active complex [Ru(η6-p-cym)(bpm)(H2O)]2+ [12]. Other authors have explored the conjugation of peptide cyclo-RGDfK with different ruthenium-polypyridyl complexes for applications in targeted therapy and/or diagnostics of human breast adenocarcinoma, glioblastoma, cervical cancer, and head and neck tumors [13][14][15]. Kühn and co-workers studied the vectorization of a terpyridine-based ruthenium complex towards ανβ3-expressing cancer cells by using one or two cyclo-RGDfK peptides [13]. Conjugates 2 and 3 (Figure 3) were synthesized via amide bond formation between the amine group present at the sidechain of the lysine residue of the targeting-peptide and the carboxylic acid group of the precursor complexes [Ru(terpy)(terpyCOOH)]2+ or [Ru(terpyCOOH)2]2+, respectively (where terpy = 2,2′:6′,2′′-terpyridine; terpyCOOH = [2,2′:6′,2′′-terpyridine]-4′-carboxylic acid). Both conjugates showed high affinity and selectivity towards the ανβ3 integrin (IC50 2 = 49 nM; IC50 3 = 2.5 nM) as compared to its αVβ5 analogue (IC50 2 > 1000 nM; IC50 3 = 595 nM). The 20-fold higher affinity displayed by the dipeptide conjugate 3 comparatively to the mono-derivatized one emphasizes the role of cyclo-RGDfK as the targeted delivery agent. However, both conjugates showed low in vitro cytotoxicity (IC50 values > 85 μM) against both cell lines with scarce expression of ανβ3 (A549, human non-small-cell lung cancer) or moderate expression of this receptor (SKOV3, human mammary carcinoma), with no significant difference between them. The poor antiproliferative activity of 2 and 3 is attributed to the intrinsic lack of cytotoxicity of the free ruthenium complexes and to their low uptake by the cancer cells despite the increased affinity of the conjugates towards the integrin receptors [13].
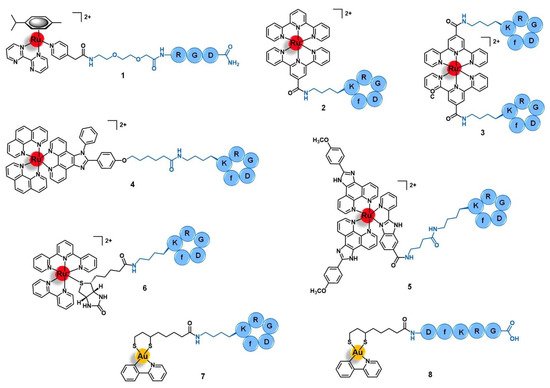
Figure 3. Ru(II)- and Au(III)-conjugates containing an integrin-targeting peptide as the delivering agent of the active metal complex into cancer cells: conjugates with the peptides RGD (1), cyclo-RGDfK (2 to 7), and DfKRG (8). Note: f = D-phenylalanine.
The photodynamic therapy (PDT) of cancer is a non-invasive approach based on the use of photoactivable sensitizers to elicit a local anti-tumor response upon specific light irradiation [14].
Aimed at the development of a new precise photodynamic therapeutic approach against human glioblastoma, Wang et al. vectorized the complex [Ru(phen)2(phenimi)]2+ (phen = 1,10-phenanthroline; and phenimi = 6-(4-(1-phenyl-1H-imidazo[4,5-f][1,10]phenanthrolin-2-yl) phenoxy)hexanoic acid) with cyclo-RGDfK (4, Figure 3) [15]. Conjugate 4 selectively targets the mitochondria of αVβ3-overexpressing glioblastoma cells and induces cell death under appropriate light irradiation conditions, both in vitro and in vivo. It preferentially accumulates in αVβ3-positive cancer cells (U87MG, human glioblastoma) rather than in αVβ3-negative cancer cells (MCF-7, human breast adenocarcinoma) with a higher degree of cellular uptake than the respective non-vectorized ruthenium complex in U87MG cells, but a similar degree of internalization in MCF-7 cells. The selective uptake of 4 in U87MG cell lines seems to be mediated by the αVβ3 integrins by blocking assays with the RGD tripeptide in this cell line. After internalization by U87MG cells, conjugate 4 selectively accumulates in the mitochondria (85%) and cytosol, being excluded from the nucleus. Moreover, contrary to the non-vectorized complex, the conjugate was shown to have a selective cytotoxic action in vitro, being more active upon irradiation than in dark conditions against the αVβ3(+) cells and without significant cytotoxicity against the αVβ3(−) cell line, either in the dark or upon irradiation. Further in vitro studies with 3D multicellular tumor spheroids of U87MG cells showed that 4 has deeper tissue penetration and was able to reduce the diameter of the spheroids over time, showing suitable characteristics for the PDT of deep tissues. Regarding the mechanism of action, in this 3D model and upon light irradiation, the conjugate induced the production of reactive oxygen species (ROS) and induced cell death by apoptosis mediated by mitochondria-dependent signaling pathways. In vivo, it showed a remarkable inhibition of tumor growth upon two-photon PDT of U87MG tumor-bearing Balb/c mice with a tumor inhibition rate of 87% compared to the non-vectorized complex (15%) in the same conditions. Without the two-photon PDT, the conjugate only showed a rate of 29%. Moreover, conjugate 4 preferentially accumulated in the tumor rather that in the main organs of the mice, in contrast with the non-vectorized complex that was found in a higher content at the liver than in the tumor. The four-fold higher accumulation of 4 in the tumor compared to the respective non-conjugated complex is consistent with the higher cellular uptake observed and might explain the high anticancer activity together with no significant damage of the remaining healthy organs. Altogether, these results suggest that conjugate 4 has favorable properties to targeted photodynamic therapy and, according to the authors, holds the potential to be further developed as a multifunctional mitochondria-targeting agent in cancer theranostics [15].
It is known that cancer tissues have a typical microenvironment around them that differs from the healthy state, including different pH, oxygen levels, redox potential, intra and extracellular enzymes, etc. Many authors have been exploring the use of drug-delivery systems responsive to cancer-dependent stimulus for a precise and controlled release of the drug into the tumor, while remaining inert during body distribution and after reaching non-tumoral organs [16]. Chen’s group reported a ruthenium-cyclo-RGDfK prodrug (5, Figure 3) that is pH-sensitive to the acidic tumor’s microenvironment (≈ 6.5 to 6.9) and that could be potentially useful as a theranostic agent against cervical cancer [17]. Conjugate 5 was prepared by a condensation reaction between the lysin residue of cyclo-RGDfK and the carboxylic acid group of the luminescent complex [Ru(POP)2(pbiz)]2+ (POP = 2-(4-methoxyphenyl)imidazo[4,5-f]1,10-phenanthroline; pbiz = 2-(pyridin-2-yl)-1H-benzo[d]imidazole-6-carboxylic acid). Conjugate 5 showed higher in vitro uptake by CaSki, SiHa, and HeLa cervical cancer cell lines via ανβ3 integrin receptor-mediated mechanism and higher cytotoxicity than the respective non-conjugated ruthenium complex, inducing cell death by apoptosis. Furthermore, it was also shown to be less cytotoxic in other cell lines with lower expression of ανβ3 integrins (e.g., MCF-7 human breast cancer cells, Ect1/E6E7 non-tumoral cervical cells, and L02 human hepatocytes) with a safety index up to five-fold higher. Conjugate 5 is stable in solutions at physiological pH (7.4) over 24 h, however, at pH < 6.8 (tumor microenvironment), it suffers hydrolysis, with substitution of the pbiz ligand by two water molecules, releasing complex [Ru(POP)2(H2O)2]2+ from the targeting peptide. This activated aqueous Ru complex exhibits a cytotoxicity of the same order of magnitude as the non-vectorized Ru complex against the same cancer cell lines, and therefore might correspond to the active drug obtained after the conjugate reaches the tumor. Additionally to these promising results, conjugate 5 also demonstrated favorable deep-red luminescent properties after one-photon and two-photon excitation, allowing the deep tissue imaging of 3D tumor spheroids of CaSki cells. The group of Chen et al. also studied the biodistribution and the potential therapeutic effect of 5 in CaSki-inoculated xenograft mice. Interestingly, 36 h after administration of 5 (4 μmol/kg) there was a selective tumor accumulation which allowed imaging it. In contrast, the non-conjugated Ru complex was distributed non-specifically through the animal, accumulating in the liver, spleen, lung, and kidney. Remarkably, after 25 days of treatment (12 doses at 4 μmol/kg), there was a considerable tumor weight reduction (74%) without the appearance of pathological damage or abnormalities of the healthy tissues. Unlike the promising results obtained with 5, the non-conjugated complex only gave a tumor reduction of 53% and led to spleen damage. In addition, after 25 days of treatment, the tumor induced liver and renal dysfunctions in the mice treated with the free complex, but those treated with the conjugate had their kidney and liver functions back to normal after the same period. Ex vivo imaging studies of cervix tumoral and non-tumoral tissue samples from 38 human patients were also performed. Unlike the non-conjugated complex, conjugate 5 was able to distinguish healthy tissues from the tumoral ones, as well as identify cervical cancer at different stages with a sensitivity of 95% and a specificity of 100%. Owing to the promising results, the authors pointed to this conjugate as a pH-responsive delivery system able to release a luminescent and cytotoxic ruthenium complex in a controlled way after activation by the acidic microenvironment of the tumor, rendering it a potential multifunctional theranostic agent for application in the precise therapy of cervical cancer [17].
The use of ruthenium conjugates for ex vivo targeted cancer diagnosis was exploited by Casini and co-workers who reported the synthesis of conjugate 6, which was obtained by conjugation of the photocleavable complex [Ru(terpy)(bipy)(D-biotin)]2+ (bipy = 2,2′-bipyridine) to cyclo-RGDfK (Figure 3). Conjugate 6 was used as a mass-tag for potential application in targeted epitope-based laser desorption ionization mass spectrometry imaging (LDI-MSI) of hypopharyngeal squamous cell carcinoma [18]. LDI-MSI is a technique based on using laser cleavable mass-tags that bind specifically and with high affinity to given moieties present in the tissues under analysis for the detection of proteins of interest, such as the αVβ3 integrins in this case. The latter shows a characteristic distribution pattern in hypopharyngeal carcinomas, compared to healthy organs, allowing the diagnostics. Conjugate 6 binds to αVβ3 integrins with high affinity (IC50 = 3.2 nM) and selectivity (IC50 for α5β1 and αVβ5 ≈ 500 nM), a characteristic that together with its photocleavable properties render the conjugate suitable as a probe for matrix-free LDI-MSI. Inside the mass spectrometer ionization chamber, conjugate 6 can be cleaved from its molecular target on the cancer tissue surface sample upon UV-light irradiation, which releases a fragment identified as [Ru(terpy)(bipy)(pyridine)-3H]+. The latter provides a fingerprint signal in the MS spectrum with specific mass and isotopic pattern distribution that allows its unambiguous identification for indirect target detection. Moreover, incubation of the cancer tissue section with 6 allowed to clearly distinguish the signal corresponding to the distribution of the mass-tag with a pattern that correlated with the distribution of the αVβ3 integrins determined by classical methods of immunohistochemistry and hematoxylin staining. On the other hand, incubation with the non-conjugated complex resulted in unspecific and scarce detection by LDI-MSI. Given these results, the authors suggested that conjugate 6 holds potential to be employed as a sensitive tool for matrix-free targeted LDSI-MSI and that further modifications of the Ru fragment and/or of the targeting moiety would eventually allow far-reaching applications in cancer diagnostics [18].
Concerning gold complexes, only a few examples of target-delivery using integrin-binding peptides have been reported. Recently, Metzler-Nolte and colleagues developed two Au(III)-peptide conjugates based on the complex [Au(ppy)(Lpa)] (ppy = 2-phenyl-pyridine; and Lpa = lipoic acid) with the cyclo-RGDfK (7, Figure 3) and the linear DfKRG peptides (8, Figure 3) [19]. The natural product Lpa was chosen as a tethering moiety given its chemical structure and for being known for its own anticancer properties. Both conjugates were prepared by firstly derivatizing the peptides with Lpa, followed by reducing Lpa’s internal disulphide bond, and then reacting it with the complex [Au(ppy)Cl2]. Conjugate 7, containing cyclo-RGDfK, showed an eight-fold increased cytotoxicity in vitro against MCF-7 and MDA-MB-231 human breast cancer cell lines compared to the parent complex [Au(ppy)Cl2]. The conjugate with the linear DfKRG peptide (8) was shown to be less cytotoxic than the cyclic analogue (ca. 18-fold less active) and even less cytotoxic than the non-vectorized gold complex (ca. 3-fold less active). These results suggest that the use of integrin-targeting peptides as carriers might be a promising approach to the targeted delivery of gold complexes into breast cancer cells [19].
Overall, despite the use of integrin-targeting vectors for the precise delivery of cytotoxic complexes of ruthenium and gold into cancer cells being still in the very beginning of preclinical evaluation, the preliminary results suggest that this might be a successful strategy for the treatment and/or diagnosis of several types of tumors, either applied in single or in combined therapy with other well-stablished approaches such as PDT.
3. Cadherins
Although less exploited than integrins, cadherins are another class of CAM that has been explored for specific delivery of anticancer agents into tumors [2]. Cadherins are calcium-dependent adhesion glycoproteins associated with the cell-to-cell adherent junctions on solid tissues, in which they mediate and regulate the reorganization of the cell cytoskeleton, intracellular signaling, and transcriptional regulation processes, as well as angiogenesis, morphogenesis and tissue growth, differentiation, and organization [20]. Cadherins are grouped into three main families—classical cadherins (type I and II), protocadherins, and atypical cadherins. The largest and most studied family, classical cadherins are highly conservative structures expressed in a tissue-specific manner and are subclassified according to the location they are typically associated with, for example, neural (N)-, epithelial (E)-, placental (P)- and vascular endothelial (VE)-cadherins [20]. Alterations in cadherin-mediated processes are often associated with cancer growth and dissemination. For instance, many tumors go through a phenomenon called cadherin switch, in which N-cadherins are upregulated while E-cadherins are downregulated, inducing tumor cells to resist natural apoptosis and gain invasive and metastatic capacity [21][22]. Consequently, E- and N-cadherins have both been exploited as targets for cancer therapy, with some drug candidates achieving clinical trials [21][22]. The overexpression of cadherins in several types of cancer cells compared to the remaining non-tumoral tissues and their role in the permeation of biological barriers through the paracellular pathway have also opened the possibility of exploring them as receptors for targeted drug delivery [21][22][23]. A common approach involves the use of antagonist peptides containing the histidine-alanine-valine (HAV) sequence, as this motif corresponds to the cell adhesion recognition sequence present in the extracellular subdomain EC1 of cadherins that is essential for their correct adhesion and function. HAV-based peptides can bind selectively and with high affinity to cadherins, thus being used either as targeted anticancer agents or targeted drug delivery carriers [22][23].
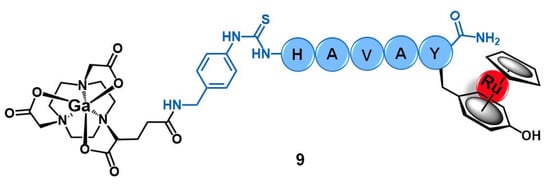
Buglyó and co-workers reported the first heterobimetallic conjugate with an HAV-based peptide for application in cancer theranostics [24]. The peptide-containing radioactive complex 67Ga-NODAGA-[(η6-Tyr-RuCp)-HAVAY-NH2] (9, Figure 4, Cp = η5-C5H5; HAVAY = his-ala-val-ala-tyr; and NODAGA = 2,2′-(7-(1-carboxy-4-((4-isothio cyanatobenzyl)amino)-4-oxobutyl)-1,4,7-triazonane-1,4-diyl)diacetic acid) was prepared by metalation of the HAVAY at its tyrosine residue with [RuCp(η6-naphthalene)] under visible-light irradiation, forming a sandwich-type Ru(II) complex, followed by conjugation to NODAGA and labeling with 67Ga(III). Conjugate 9 was designed aiming to contain three functional moieties for targeted theranostics: i) the HAVAY peptide to target the cadherins overexpressed at cancer cells and thus acting as a drug carrier, ii) a Ru(II) complex with potential anticancer activity, and iii) a 67Ga-radiolabeled core (γ-emitter) for imaging purposes by single photon emission computed tomography (SPECT). The cellular uptake of the conjugate was determined in four human cancer cell lines with different expression levels of N-/E-cadherins, namely A375(+/−) melanoma, PC-3(+/+) prostate, and MCF-7(−/+) and MDA-MB-231(−/−) breast cancer cells. Surprisingly, conjugate 9 showed low to moderate uptake, not related to the cadherins expression levels, with the highest uptake rate found in MDA-MB-231 cells (14.9%) in which the conjugate was mostly retained at the cell membrane. Additionally, in this cell line, the conjugate also was not shown to be cytotoxic, most probably due to the low cellular internalization and retention [24].
Even though cadherins are still sparsely exploited as targets for the specific delivery of ruthenium and gold complexes into cancer cells, with plenty of room for further studies aiming to optimize and take full advantage of this approach, we believe that it might become a promising strategy given the good results found for other classes of cell-adhesion molecules (such as integrins, see previous section).
References
- Harjunpää, H.; Asens, M.L.; Guenther, C.; Fagerholm, S.C. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front. Immunol. 2019, 10, 1078.
- Dunehoo, A.L.; Anderson, M.; Majumdar, S.; Kobayashi, N.; Berkland, C.; Siahaan, T.J. Cell Adhesion Molecules for Targeted Drug Delivery. J. Pharm. Sci. 2006, 95, 1856–1872.
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300.
- Bachmann, M.; Kukkurainen, S.; Hytönen, V.P.; Wehrle-Haller, B. Cell adhesion by integrins. Physiol. Rev. 2019, 99, 1655–1699.
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367.
- Millard, M.; Odde, S.; Neamati, N. Integrin Targeted Therapeutics. Theranostics 2012, 1, 154–188.
- Marsico, G.; Russo, L.; Quondamatteo, F.; Pandit, A. Glycosylation and Integrin Regulation in Cancer. Trends Cancer 2018, 4, 537–552.
- Temming, K.; Schiffelers, R.M.; Molema, G.; Kok, R.J. RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug Resist. Updat. 2005, 8, 381–402.
- Katsamakas, S.; Chatzisideri, T.; Thysiadis, S.; Sarli, V. RGD-mediated delivery of small-molecule drugs. Future Med. Chem. 2017, 9, 579–604.
- Oliveira, M.C.; Correia, J.D.G. Biomedical applications of radioiodinated peptides. Eur. J. Med. Chem. 2019, 179, 56–77.
- Correia, J.D.G.; Paulo, A.; Raposinho, P.D.; Santos, I. Radiometallated peptides for molecular imaging and targeted therapy. Dalt. Trans. 2011, 40, 6144–6167.
- Barragán, F.; López-Senín, P.; Salassa, L.; Betanzos-Lara, S.; Habtemariam, A.; Moreno, V.; Sadler, P.J.; Marchán, V. Photocontrolled DNA binding of a receptor-targeted organometallic ruthenium(II) complex. J. Am. Chem. Soc. 2011, 133, 14098–14108.
- Hahn, E.M.; Estrada-Ortiz, N.; Han, J.; Ferreira, V.F.C.; Kapp, T.G.; Correia, J.D.G.; Casini, A.; Kühn, F.E. Functionalization of Ruthenium(II) Terpyridine Complexes with Cyclic RGD Peptides To Target Integrin Receptors in Cancer Cells. Eur. J. Inorg. Chem. 2017, 2017, 1667–1672.
- Hu, J.J.; Lei, Q.; Zhang, X.Z. Recent advances in photonanomedicines for enhanced cancer photodynamic therapy. Prog. Mater. Sci. 2020, 114, 100685.
- Zhao, Z.; Qiu, K.; Liu, J.; Hao, X.; Wang, J. Two-photon photodynamic ablation of tumour cells using an RGD peptide-conjugated ruthenium(ii) photosensitiser. Chem. Commun. 2020, 56, 12542–12545.
- He, Q.; Chen, J.; Yan, J.; Cai, S.; Xiong, H.; Liu, Y.; Peng, D.; Mo, M.; Liu, Z. Tumor microenvironment responsive drug delivery systems. Asian J. Pharm. Sci. 2020, 15, 416–448.
- Zhao, Z.; Zhang, X.; Li, C.; Chen, T. Designing luminescent ruthenium prodrug for precise cancer therapy and rapid clinical diagnosis. Biomaterials 2019, 192, 579–589.
- Han, J.; Sun, J.; Song, S.; Beljaars, L.; Groothuis, G.M.M.; Permentier, H.; Bischoff, R.; Halmos, G.B.; Verhoeven, C.J.; Amstalden van Hove, E.R.; et al. Targeted imaging of integrins in cancer tissues using photocleavable Ru(ii) polypyridine complexes as mass-tags. Chem. Commun. 2020, 56, 5941–5944.
- Śmiłowicz, D.; Slootweg, J.C.; Metzler-Nolte, N. Bioconjugation of Cyclometalated Gold(III) Lipoic Acid Fragments to Linear and Cyclic Breast Cancer Targeting Peptides. Mol. Pharm. 2019, 16, 4572–4581.
- Gloushankova, N.A.; Rubtsova, S.N.; Zhitnyak, I.Y. Cadherin-mediated cell-cell interactions in normal and cancer cells. Tissue Barriers 2017, 5.
- Wong, S.H.M.; Fang, C.M.; Chuah, L.H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018, 121, 11–22.
- Blaschuk, O.W. N-cadherin antagonists as oncology therapeutics. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370.
- On, N.H.; Kiptoo, P.; Siahaan, T.J.; Miller, D.W. Modulation of blood-brain barrier permeability in mice using synthetic E-cadherin peptide. Mol. Pharm. 2014, 11, 974–981.
- Bihari, Z.; Vultos, F.; Fernandes, C.; Gano, L.; Santos, I.; Correia, J.D.G.; Buglyó, P. Synthesis, characterization and biological evaluation of a 67Ga-labeled (η6-Tyr)Ru(η5-Cp) peptide complex with the HAV motif. J. Inorg. Biochem. 2016, 160, 189–197.



