3.1. Clinical Characteristics
NF2 is an autosomal dominantly inherited syndrome that predisposes individuals to multiple nervous tumors. A de novo mutation may take place after fertilization, resulting in a mosaic expression
[49][50]. Diagnosis is based on clinical and neuroimaging studies (). Two large population-based studies reported that this condition occurs in 1 in 25,000 people
[51]. The actuarial survival after diagnosis is 15 years, with an average age at death of 36 years
[52] and a 10-year survival rate of 67%
[53]. NF2 patients uniformly develop schwannomas on the bilateral vestibular portion of the eighth cranial nerve and on other cranial nerves, spinal roots, or peripheral nerves
[54]. In addition, NF2 patients often develop multiple meningiomas and ependymomas at an early age.
Table 3. Diagnostic criteria of neurofibromatosis type 2.
NF2 patients often experience hearing loss, balance problems, flesh colored skin flaps, and muscle wasting. Some develop mononeuropathy, often involving the facial nerve. Severe polyneuropathy is noted in 3–5% of adult NF2 patients
[55]. Visual impairment is likely due to cataracts, optic nerve meningiomas, and retinal hamartomas
[56][57][58]. Approximately 70% of NF2 patients have cutaneous manifestations: only 10% have more than 10 skin tumors
[54]. Plaque-like lesions may be more pigmented than the surrounding skin with increased hair. Subcutaneous nodules are identified along the peripheral nerve. Intracutaneous schwannomas, which are similar to those observed in NF1 patients, are occasionally seen.
3.2. Genetic and Molecular Characteristics
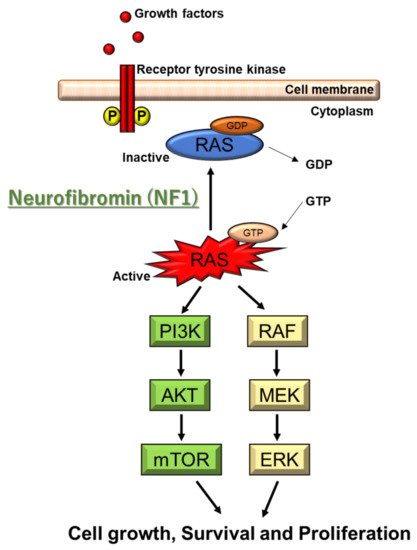
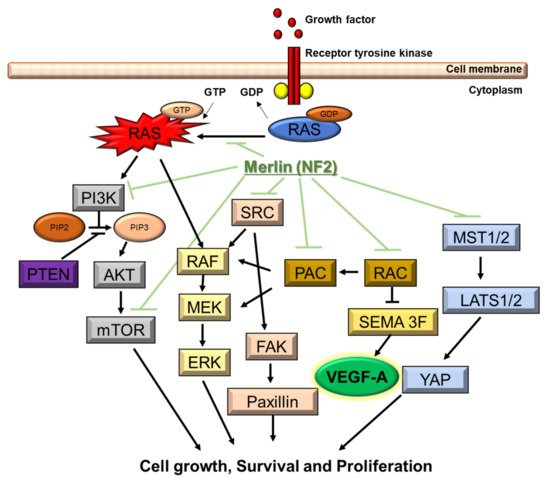
NF2 is caused by a defect in the gene that normally produces merlin, located at 22q12.2 of chromosome 22, which regulates multiple proliferative signaling pathways. At the membrane, merlin blocks signaling caused by integrins and tyrosine receptor kinases. Merlin can also inhibit downstream signalings, including the p21-activated kinase signaling, Ras/Raf/MEK/ERK, FAK/Src, PI3K/AKT, Rac/PAK/JNK, mTORC1, and Wnt/β-catenin pathways (). In the nucleus, merlin suppresses the E3 ubiquitin ligase CRL4DCAF1, which also regulates the expression of integrins and tyrosine receptor kinases. The Hippo signaling pathway regulates tissue homeostasis. Merlin is implicated as one of the upstream regulators of the Hippo signaling pathway
[49][50][59][60][61].
Figure 2. Molecular pathogenesis of NF2. NF2 gene encodes merlin. Merlin regulates multiple proliferative signaling pathways. At the membrane, merlin blocks signaling caused by integrins and tyrosine receptor kinases. Merlin can also inhibit downstream signalings, including the p21-activated kinase signaling, Ras/Raf/MEK/ERK, FAK/Src, PI3K/AKT, Rac/PAK/JNK, mTORC1, and Wnt/β-catenin pathways. Downstream signaling of NF2 includes VEGF-A.
A de novo mutation results in a mosaic expression. Somatic mosaicism may prevent the molecular diagnosis unless tumor tissue is analyzed
[62]. The growth of schwannomas requires inactivation of both NF2 alleles. The “second hit” occurs through loss of the entire NF2 gene and most of chromosome 22. Various types of mutations, such as protein-truncating alterations (frameshift deletions/insertions and nonsense mutations), splice-site mutations, missense mutations, are identified. Truncating mutations (nonsense and frameshifts) are the most frequent germline event and cause the most severe disease. The presence of a truncated protein is associated with younger age at diagnosis and a higher prevalence of meningiomas, spinal tumors, and cranial nerve tumors other than VIII
[62]. Deletions in the NH2-terminal domain of merlin proteins are associated with early tumor onset and disease progression
[62]. The most common alterations are splice-site mutations or nonsense mutations in exons 1–8. Missense or in-frame deletions have been associated with milder clinical courses. A positional effect with mutations in the latter parts of the gene (exons 14 and 15) is associated with milder disease and fewer meningiomas
[63]. Alterations in the conserved N-terminal FERM domain and truncating mutations are typically associated with the Wishart phenotype including younger age at diagnosis, a higher incidence of meningiomas, ophthalmologic and cutaneous lesions, and poor outcomes. Missense and splice-site mutations, particularly in the 3′ end of the gene, are associated with the Gardner phenotype and a better prognosis with fewer meningiomas
[64].
The role of mutation screening for NF2 in all patients with a unilateral vestibular schwannoma is less certain. Although these patients show an increased risk for the development of NF2, routine screening for germline mutations is not recommended except in patients younger than 30 years
[65][66], but can undergo prenatal diagnosis and pre-implantation genetic diagnosis.
3.3. Therapeutic Strategies
To date, there is no established effective treatment for NF2 patients because tumors are highly likely to regrow after surgical resection
[67]. Treatment is generally indicated when the patient has risk of brainstem compression, deterioration of hearing, and/or facial nerve dysfunction. Vestibular schwannomas may involve facial nerve fibers, possibly posing a significant risk of damage to the facial nerve during surgery
[68]. While the use of stereotactic radiosurgery has recently become an effective management modality for NF2 schwannomas, it is not advocated for multiple or large tumors
[69]. Vestibular dysfunction and trigeminal neuropathy have been reported after radiosurgery
[70]. Malignant transformation associated with radiosurgery is evidently uncommon
[71][72]. Furthermore, surgical resection may be more difficult following stereotactic radiosurgery
[69][73].
Vascular endothelial growth factor (VEGF)-A is an important factor for the growth of schwannoma that mainly depends on VEGF-A/VEGF receptor (VEGFR) pathway (not other factors such as estrogen and progesterone)
[74][75]. Tumor shrinkage and hearing improvement are identified after administration of bevacizumab (a monoclonal antibody against VEGF-A) in >50% of progressive vestibular schwannomas in NF2 patients. Stable hearing is retained in the majority of the patients
[76][77][78][79][80][81][82][83][84]. Bevacizumab is recently considered as a first-line medical therapy for rapidly growing vestibular schwannomas
[85]. In a recent meta-analysis of eight observational studies involving 161 patients with NF2-associated vestibular schwannomas, the best response to bevacizumab was partial regression in 41%, no change in 47%, and progression in 7% of the patients
[86]. The median treatment duration was 16 months. Hearing improved in 20%, remained stable in 69%, and worsened in 6 percent. The incidence of serious toxicity was 17%, with amenorrhea (70%), proteinuria (43%), and hypertension (33%). Although the dose and schedule of bevacizumab has not been standardized, a proposed regimen is 5–7.5 mg/kg every 2–3 weeks for at least 6 months, followed by maintenance therapy at 2.5–5 mg/kg every 4 weeks
[85]. A higher dose regimen (10 mg/kg every 2 weeks for 6 months) offered no clear advantage compared with lower-dose regimens
[87], and furthermore higher dose may increase the risk of renal impairment.
Contrarily, problems arise after administration of bevacizumab such as the need for frequent administration, occurrence of hypertension and thrombosis, and apparent drug resistance. Tumor growth rebound following bevacizumab treatment discontinuation was pointed out
[88]. Recently, a clinical trial using VEGFR1/2 peptide vaccine was also conducted in patients with progressive NF2-derived schwannomas, showing hearing improvement and tumor volume reduction
[89].
The NF2 gene product involves multiple molecular pathways in cell growth. Some studies reported on the efficacy of everolimus, an oral inhibitor of the mTORC1, for progressive vestibular schwannomas in NF2 patients
[90][91]. Lapatinib showed objective activity in 4 out of 17 patients with NF2-related progressive vestibular schwannoma in a phase II trial
[92], whereas erlotinib was not effective in a retrospective series with 11 NF2 patients
[93]. Mirdametinib (PD-0325901) is an orally delivered inhibitor of the dual specificity kinases (MEK1 and MEK2), which demonstrated tumor shrinkage and sustained inhibition of pERK. Furthermore, the recent study demonstrated that hypoxia was significantly associated with shorter progression-free survival in NF2 schwannomas
[94]. HIF-1-targeted therapy might be considered for some NF2 schwannomas that are difficult to treat by surgical resection and stereotactic radiosurgery
[94].
For patients with severe hearing impairment, strategies using cochlear or brainstem implants may offer some benefit
[95][96]. Pilot studies have demonstrated the feasibility of novel psychosocial interventions delivered to deaf individuals with NF2 via teleconferencing with captioning technology
[97].
NF2 patients tend to develop meningiomas at an earlier age than those with sporadic meningiomas
[98]. The meningiomas seen in NF2 patients are more frequently atypical or anaplastic compared with sporadic tumors
[99]. Although radiation therapy has been performed in those patients, long-term follow-up is lacking. Targeted therapies are under investigation
[100]. Lapatinib showed some activity in a small number of patients with NF2-associated progressive meningiomas
[101]. There is little evidence that bevacizumab has activity in NF2-related meningiomas
[102]. These molecular studies led to clinical trials using mTORC1 inhibitor everolimus, a rapamycin analog, for NF2 and sporadic meningiomas. Alternate treatment options for NF2 tumors include inhibitors of the epidermal growth factor receptor, an inhibitor of platelet-derived growth factor, and an inhibitor of histone deacetylase
[100].
3.5. Animal Models
In the Nf2 mutant allele, the 3′ part of exon 2 up to the 5′ part of intron 3 has been replaced by a selection marker
[104]. Two different mutant Nf2 alleles were generated
[105]. One mutant allele, Nf2
KO3, was generated by an insertional mutation in exon 3. The other mutant allele, Nf2
Δ2, carried an in-frame deletion of exon 2. Tissue-specific Nf2 inactivation can be accomplished by the Cre/LoxP technology. LoxP recombinatorial sequences are inserted into noncoding regions flanking exon 2 of the Nf2 gene to produce phenotypically normal Nf2 flox mice. Tissue-specific inactivation is mediated by the expression of the bacteriophage Cre recombinase from a tissue-specific promoter or by direct injection of adenoviral Cre
[106].
Recently, Chen et al. described the injection of schwannoma cells into the mouse brain cerebellopontine angle region and the application intravital imaging and hearing assessment techniques to study tumor growth and hearing loss. In addition, ataxia, angiogenesis, and tumor–stroma interaction assays could be shown
[107].
To date, models of NF2-associated ependymoma remain yet to be generated whereas genetically engineered mouse strain of meningiomas have been. Somatic Nf2 loss after subarachnoid or subdural viral injection of Cre recombinase into newborn Nf2 flox/flox conditional knockout mice result in meningioma
[106]. More aggressive tumors develop when NF2 loss is coupled with the loss of other tumor suppressor genes (Ink4a)
[108]. It has been challenging to maintain NF-associated tumors as patient-derived xenografts. To date, successful orthotopic transplant models have only been developed for NF2-associated meningioma
[109].