Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marina Barcena-Varela | + 3090 word(s) | 3090 | 2021-06-02 10:49:01 | | | |
| 2 | Peter Tang | Meta information modification | 3090 | 2021-06-03 08:28:56 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Barcena-Varela, M. Hepatocellular Carcinoma Heterogeneity. Encyclopedia. Available online: https://encyclopedia.pub/entry/10438 (accessed on 08 February 2026).
Barcena-Varela M. Hepatocellular Carcinoma Heterogeneity. Encyclopedia. Available at: https://encyclopedia.pub/entry/10438. Accessed February 08, 2026.
Barcena-Varela, Marina. "Hepatocellular Carcinoma Heterogeneity" Encyclopedia, https://encyclopedia.pub/entry/10438 (accessed February 08, 2026).
Barcena-Varela, M. (2021, June 02). Hepatocellular Carcinoma Heterogeneity. In Encyclopedia. https://encyclopedia.pub/entry/10438
Barcena-Varela, Marina. "Hepatocellular Carcinoma Heterogeneity." Encyclopedia. Web. 02 June, 2021.
Copy Citation
Tumor heterogeneity in liver cancer is a major contributor to the high lethality rate found in patients suffering from this disease. The therapeutic outcomes are drastically affected by this heterogeneity, which complicates patient stratification and response prediction.
hepatocellular carcinoma
heterogeneity
molecular mechanisms
risk factors
microenvironment
1. Introduction
Liver cancer represents a major health problem, causing more than 800,000 deaths annually worldwide [1]. The most frequent type of liver cancer, which represents the second leading cause of cancer-related mortality, is hepatocellular carcinoma (HCC) [2][3]. Although HCC treatment has greatly improved over the past decades, HCC patients are generally diagnosed at advanced stages and ineligible for curative ablative therapies. There are several non-surgical therapeutic options available to treat HCC such as chemoembolization, radiofrequency ablation, or radiation therapy. However, liver resection and liver transplantation remain the main curative therapy options [4][5][6]. Concerning systemic treatment options, there are several therapies approved for advanced HCC patients, including the multikinase inhibitors sorafenib, regorafenib, lenvatinib, carbozantinib; the anti-angiogenic antibody ramucirumab; the immune checkpoint inhibitors nivolumab and pembrolizumab; and the combination therapies atezolizumab (anti-PD-L1) + bevacizumab (anti-VEGFA), and nivolumab (anti-PD-1) + ipilimumab (anti-CTLA4) [7][8][9][10][11][12][13]. Although these agents improve the survival of patients, with objective response rates of around 30% and survival benefits of several months, there is still room for substantial clinical improvements. One plausible explanation for the unsatisfactory clinical results is the high inter-patient heterogeneity among HCC patients and the lack of validated biomarkers that can help select the patients that are most likely to respond to each therapy.
In addition to the vast inter-patient heterogeneity, HCCs are characterized by high levels of intra-tumor heterogeneity, which entail that tumor cells within a tumor are phenotypically different [14][15]. Moreover, approximately 70% of HCC patients develop early-stage recurrence following curative resection [16][17] and many studies have revealed that an elevated proportion of these recurrent tumors harbor clones that are different from the primary tumor, indicating de novo development or synchronous tumors rather than residual tumor or intrahepatic micrometastasis, contributing to intra-patient tumor heterogeneity within a single patient [14][18][19]. Most importantly, the unique etiology of HCC, associated with liver damage in 90% of all the cases, gives rise to high extra-tumor heterogeneity associated with the specific liver damaging mechanism and the host microbiome and immune system.
2. Inter-Patient Heterogeneity in HCC
The high inter-patient heterogeneity in HCC limits the availability of predictive biomarkers, complicates the development and clinical implantation of targeted therapies, and contributes to the poor outcome of the patients. The dissection of the contribution of each tumor-extrinsic and tumor-intrinsic factor in the process of hepatocarcinogenesis may help in the future to discover novel biomarkers and better define prognosis in patients.
2.1. Diversity of Tumor-Extrinsic Factors: Extra-Tumor Heterogeneity
HCC is a complex malignancy that can be triggered by many different factors including hepatic viral infections, alcohol abuse, and diet-induced metabolic disorders. The different etiologies can promote various oncogenic pathways and most liver tumors (up to 90%) develop in the context of underlying chronic liver disease (CLD) that is characterized by the accumulation of extracellular matrix (fibrosis), scar formation (fibrosis and cirrhosis), and chronic inflammation, which together constitute the perfect pro-oncogenic niche for tumor initiation and progression [19][20][21].
More importantly, the surrounding microenvironment greatly affects the phenotypic output of liver tumors, even of those presenting activation of the same oncogenic pathways, contributing to HCC heterogeneity and highlighting the relevance of all the factors involved in the malignant transformation of hepatocytes [22]. Microenvironmental factors, such as the host gut microbiota, have also been shown to play an essential role in liver tumorigenesis, and the precise mechanisms of this contribution are starting to be elucidated [23][24].
Among the different tumor-extrinsic factors, demography is a very relevant epidemiologic aspect to consider. Viral infections caused by hepatitis B virus (HBV) and environmental exposure to aflatoxin B1, for example, are the most frequent risk factors in Asia and Africa, whereas hepatitis C virus (HCV) infection, chronic alcohol intake, and metabolic syndrome are frequently related to HCC in Western countries [17][25]. Among the different hepatitis viruses, only hepatitis B and C viruses (HBV, HCV) have been demonstrated to cause HCC [26], whereas hepatitis D viruses (HDV) can be found in HCC patients that are co-infected with HBV. However, the contribution of HDV to the carcinogenic process is still not clear [27][28]. HBV infection presents its highest prevalence in Asia followed by Africa, while the epidemiological distribution of HCV infection is more globally spread, with Japan, Italy, and the United States among the countries with the highest prevalence [29]. The precise oncogenic mechanisms driven by hepatitis viral infections are not completely understood. Myriad molecular and cellular events related to viral injection have been characterized including intracellular DNA rearrangement and chromatin instability, promotion of mutations or overexpression by DNA integration (insertional mutagenesis), stimulation of proliferation, and activation of inflammatory signals leading to chronic inflammation within the liver. This persistent hepatocyte inflammation is one of the main contributors to premalignant stages in the liver [30]. Additionally, viral infections by HBV and HCV have been associated with immune escape and exhaustion of virus-specific T-cells as a result of sustained antigenic stimulation [31][32], shaping a liver immune microenvironment that can greatly influence tumor development and progression.
The exposure to environmental carcinogens has also been linked to the emergence of specific genetic alterations. The best known example, aflatoxin B1 (AFB1), is a mycotoxin that has been found in contaminated food, mainly in Asia and Africa, and is associated with a specific genetic signature of HCC characterized by a high rate of C > A mutations and the R249S specific mutation in the tumor suppressor TP53 [33][34]. In addition, single nucleotide polymorphisms (SNPs) of glutathione s-transferase enzyme gene variations GTSM1 and GSTT1, genes involved in carcinogen detoxification, are associated with a high risk of HCC in patients exposed to different genotoxic contaminants [35][36].
2.2. Tumor-Intrinsic Factors: The Genetic and Epigenetic Landscape of HCC
During hepatocarcinogenesis, as well as during the carcinogenic process of other solid tumors, numerous genetic alterations emerge and accumulate during the different stages of the disease, resulting in unique cancer genomes. An accurate landscape of the genetic alterations in HCC has been established thanks to the cumulative data from high-throughput sequencing efforts for large numbers of samples from patients. In order to improve the understanding of the molecular basis of HCC, several molecular subclasses presenting distinct oncogene signaling pathways and recurrent mutations have been established [22][37][38][39][40][41].
Unlike other tumor types, HCC rarely appears as a consequence of monogenic syndromes. Exceptionally, HCC has been seen in patients with APC germ-line mutations or presenting rare monogenic metabolic diseases such as hemochromatosis (HFE1 gene alterations), Wilson disease (ATP7B gene alterations), tyrosinemia type I (FAH gene alterations), cutanea tarda (UROD gene alterations), a1 antitrypsin disease (SERPINA1 gene alterations), von Gierke disease (G6PC gene alterations), or diabetes of the young type 3 (MODY3 or hepatocyte nuclear factor 1A; HNF1A gene alterations) [22][42]. In addition to these rare cases of monogenic-disease-induced HCC, hepatocellular adenoma (HCA) is a rare benign tumor developed from hepatocytes in the context of a non-damaged normal liver. These tumors, extensively studied by the laboratory of Prof. Zucman-Rossi, are associated with pre-menopausal young women, linked with specific genetic signatures, and divided into three molecular subclasses defined by (a) inactivation of the transcription factor HNF1A, (b) activation of the WNT/β-catenin pathways by CTNNB1 mutations, or (c) activation of the IL6/STAT3 pathway by somatic mutation of IL6ST, GNAS, or STAT3 [39].
Most of the polymorphisms associated with HCC development are related to specific etiologies in the context of CLD, highlighting the close relation between the different environmental factors and the genetic background in the liver [22]. Although it is hard to determine the dominant reprogramming events involved in hepatocarcinogenesis, there are some clear examples of these (micro) environmental–genetic correlations. HBV infection, for example, induces chromosome instability and insertional mutagenesis [41][43] and has been associated with TP53 mutations [37], whereas patatin-like phospholipase domain-containing protein 3 (PNPLA3) polymorphisms (coding a lipase involved in triacylglycerol hydrolysis) are strongly associated with the chronic liver disorders ALD [44] and NAFLD [45][46]. Transmembrane 6 superfamily member 2 (TM6SF2) variants are also strongly associated with the presence of NAFLD and have been correlated with the severity of steatosis, NASH, and cirrhosis [47][48]. Other examples are mutations in CTNNB1 gene encoding for β-catenin protein, which have been correlated with alcoholic HCC [37]. In addition to differences based on different genetic alterations, the sequential appearance of the genetic alterations can be different depending on the surrounding etiology of HCC and the tumor stage. In normal hepatocytes undergoing hyperproliferation (HCA), CTNNB1 mutations usually occur prior to the telomerase reverse transcriptase (TERT) promoter mutations, whereas in the case of HCC development in cirrhotic liver (accounting for most of the cases), TERT promoter mutations are observed first, in pre-cancerous stages [22][37][49]. However, alterations in some fibroblast growth factor family genes such as FGF3, FGF4, FGF19/CCND1 amplification, and TP53 and Cyclin Dependent Kinase Inhibitor 2A (CDKN2A) mutations and deletions, appeared at more advanced stages [37]. Despite the identified risk-factor-specific mutational profile, there is still tremendous heterogeneity within each etiological group and the high diversity of the altered pathways found in HCC still hinders the stratification of the patients based on their molecular signature.
2.3. Tumor-Intrinsic Factors: Molecular Subclasses in HCC
As explained above, genetic alterations in HCC are found in a variety of cancer driver pathways with a highly heterogeneous pattern of alteration among patients [37][50][51]. The main challenge is to classify the different molecular profiles of HCC, in order to stratify the patients and more accurately define the treatment options and prognosis. However, subclassifications based only on genomic profiling do not encompass all the factors that contribute to the phenotypic heterogeneity observed in tumors, either because they do not integrate other factors, such as tumor-extrinsic factors, or due to the complexity and contribution of the possible combinations of mutations found in this cancer. There are 2 major subgroups of HCC tumors accurately defined by different biological processes: the proliferation and non-proliferation subclasses [52]. Although this classification is mainly based on the gene expression profile of the tumors, it is strongly associated with clinical and pathological features of the patients and their surrounding etiology, thus integrating different sources of information to define clinical outcomes [22]. On one hand, the proliferation subclass accounting for around 50% of patients is enriched in signals related to cell proliferation and cell cycle progression and is generally associated with a more aggressive phenotype and poor outcome. The activated signaling cascades found in this group and related to proliferation and cell survival include MET or hepatocyte growth factor receptor (HGFR) pathway [53], TGFβ signaling, insulin-like factor I (IGF) pathway [54], RAS/mitogen-activated protein kinase pathways [55], and AKT/MTOR signaling pathway [56], among others. Enrichment in the expression patterns associated with stem cell features (e.g., NOTCH) [57] and markers of progenitor cells (e.g., epithelial cell adhesion molecule) [58], also found in this subclass, indicate a moderate/poor cell differentiation state that correlates with more aggressive clinical behavior [59][60]. Another characteristic aspect of this subgroup is that tumors present higher rates of chromosomal instability due to aberrant epigenetic changes. In fact, there is a defined DNA methylation-based prognostic signature [61] that correlates with signatures of progenitor cells and poor survival. On the other hand, the non-proliferation subclass is characterized by a well-differentiated phenotype, with normal hepatocyte-like features. This subgroup of HCC is less aggressive and correlates with low levels of a-fetoprotein (AFP), the most widely used biomarker to detect HCC, contrary to the high levels found in the proliferation subclassified patients [22]. One subset of this group is characterized by the liver-specific activation of WNT signaling pathway (about 25%), mainly by mutations in CTNNB1 [62], while the other main subgroup is associated with immune response [63]. Overexpression of the epidermal growth factor receptor (EGFR) is also observed in the non-proliferation group and plays a critical role in the maintenance of the transformed phenotype of HCC cells [64]. Regarding etiologic factors, HBV-related HCC tumors are predominantly classified in the proliferation subclass, whereas HCV and alcohol-related HCC are more prevalent in the non-proliferation subclass.
3. Intra-Tumor and Intra-Patient Heterogeneity
Intra-tumor heterogeneity is a feature frequently found in many solid tumors [65][66]. HCCs are frequently detected with a nodule-in-nodule appearance, where a subclone resides embedded in another [67][68]. Many other patients present multiple hepatic tumors that can develop from different clones (presenting more than one primary tumor simultaneously) or arise from a single original tumor via intrahepatic metastasis [69][70]. Tumor cells are subjected to a natural selection process within the tumors and the microenvironment that leads to clonal evolution and the coexistence of diverse clones in a single tumor. It is commonly thought that this intratumor heterogeneity is critically associated with the development of resistance against cancer therapies. In this sense, it can be hypothesized that a therapy might have an anti-tumor effect globally while inducing a positive selection in the most resistant tumoral subclones.
About 70% of the patients with HCC under curative treatment options develop recurrence, considered early-recurrence within two years of treatment or late-recurrence when it occurs after more than two years [16][17][71][72]. Although there are some studies indicating that genetic and DNA methylation patterns of HCC can be used to identify patients at high risk for recurrence [18], the origin and evolution of the relapsed tumors are still controversial. Traditionally, it was considered that early-relapsed tumors were derived from residual or intrahepatic micrometastasis rather than de novo tumorigenesis or synchronous tumors with variable clonality. However, in a study from 1989 [14] it was already shown that there is not a clear correlation between the time of appearance of the recurrence and the clonality of the recurrent tumors, an observation that was later supported by other researchers [18][73], that there is not a clear correlation between the time of appearance of the recurrence and the clonality of the recurrent tumors. More recently, an exhaustive clonality analysis at single-cell level of early relapsed HCC cohorts demonstrated that tumoral cells in relapsed tumors derived from a minor clone from the primary tumor and were characterized by a distinct tumor microenvironment that conferred enhanced immune evasion capabilities [73]. Focusing on evolutionary trajectories in liver cancer, Ding and colleagues [18] identified TP53, CTNNB1, and TERT mutations as founder drivers and convergent mutations in key drivers including TP53, TERT, CTNNB1, TSC2, JAK1, NOTCH1, FGFR3, ATRX, and RPS6KA3. These convergent mutations can favor each ancestor clone to experience parallel series of expansions, inferring an environmental selection that allows parallel or selective seeding of ancestor clones. These findings were in line with a previous work by Torrecilla S. et al. [74] where they described TERT, TP53, and CTNNB1 mutations as trunk alterations in HCC, meaning they are ubiquitously present across different regions of the same tumor and between primary and metastatic tumors in more than 85% of the cases studied. Ding et al. [18] further defined a co-evolutionary relationship between the genome and epigenome in HCC, highlighting a close interplay between genetic and epigenetic landscapes during malignant progression and pointing towards the potential of epigenetic signatures to define novel biomarkers to trace tumor evolution and progression. Recently, Losic and colleagues characterized the interactions of different components of the HCC ecosystem during cancer evolution by integrating RNA-seq, DNA-seq, TCR-seq, and SNP information from a dataset of multiregional samples from HCC patients. Their findings indicated that clonal evolution depends on different selection pressures that are primarily immune- and treatment-mediated [75], supporting the relevant role of the tumor microenvironment in clonal selection and intra-patient heterogeneity.
From a clinical perspective, it would be expected that high intra-tumoral heterogeneity and clonal diversity in HCC correlates with worse patient prognosis and limited therapeutic options, as this enhanced heterogeneity in the tumors would trigger worse therapeutic responses in addition to more susceptibility to developing targeted therapy resistance. In fact, it has been observed that those HCC patients with higher heterogeneity correlate with a worse prognosis and poor survival [75][76]; however, additional studies are necessary to decipher this association. Most importantly, both intra-tumor and intra-patient tumor heterogeneity contribute to vastly increasing inter-patient heterogeneity.
4. Clinical Implications of HCC Heterogeneity
References to the clinical implications of liver tumor heterogeneity have been mentioned along the different aspects addressed in this revision. The devastating concerns associated with the inefficient therapeutic responses against liver cancer reaffirm the imperative need to study in detail the mechanisms driving heterogeneity, to standardize a better subclassification of the patients for clinical management. Regarding decisions about the treatment of patients with HCC, physicians commonly rely on the stage of the disease rather than its molecular singularities. However, as it has been extensively explained here, the phenotypic differences at the molecular level between patients within the same stage are abysmal. Characterization of molecular subtypes and/or oncogenic signatures have generally improved the patient outcome for several types of cancer. However, the molecular classification of HCC has not been implemented for managing patients. Due to this complexity, there are not clear inter-patient biomarkers that can predict the response to a specific therapy (with the exception of ramucirumab and high AFP levels), whereas the high incidence of intra-tumor heterogeneity can induce a positive selection in the tumor subclones that is hard to foretell. In this sense, the widespread implementation of transcriptomic profiling of tumors at single-cell resolution by performing single-cell RNA and DNA sequencing along with single-cell proteomic studies might provide a comprehensive view of the tumor microenvironment and tumor cell trajectories to more accurately provide stratification strategies for HCC patients. The implementation of this novel methodology along with the use of experimental systems to properly model and understand HCC heterogeneity will enable better therapeutic approaches, based on the identification of molecular signatures that foster the choice of one therapy over another (see Figure 1). Moreover, identification of high intra-tumor heterogeneity in patients could be decisive for the election of combinatorial therapies over single-based treatment options in order to avoid the emergence of resistance. Regarding tumor recurrence of HCC, the effects of therapeutic options can be critical as patients might have very different prognoses and/or can benefit from different therapeutic approaches; unfortunately, primary tumors are characterized in most circumstances. In fact, it might be necessary to re-biopsy recurrences, regardless of relapsed time of the patients, to establish clonal origin and make decisions about new potential therapies that might be very different from the therapies proposed in the first instance.
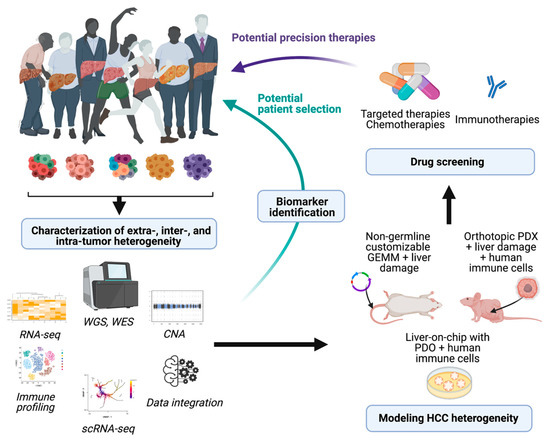
Figure 1. Potential implications of dissecting HCC heterogeneity. Implementation of high-throughput analysis to characterize HCC heterogeneity can provide useful biomarkers and pave the way for the development of novel models of HCC. The derived animal models and in vitro screenings that accurately recapitulate the complexity of this disease might represent powerful tools to test different drug sensitivities and potentially impact on the clinical management of patients. WGS, whole genome sequencing: WES, whole exome sequencing; CNA, copy number alteration; scRNA-seq, single-cell RNA-seq; PDX, patient-derived xenografts; PDO, patient-derived organoids. Figure prepared with Biorender.
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. S1), 4–13.
- Centonze, L.; Di Sandro, S.; Lauterio, A.; De Carlis, R.; Frassoni, S.; Rampoldi, A.; Tuscano, B.; Bagnardi, V.; Vanzulli, A.; De Carlis, L. Surgical Resection vs. Percutaneous Ablation for Single Hepatocellular Carcinoma: Exploring the Impact of Li-RADS Classification on Oncological Outcomes. Cancers 2021, 13, 1671.
- Di Sandro, S.; Bagnardi, V.; Cucchetti, A.; Lauterio, A.; De Carlis, R.; Benuzzi, L.; Danieli, M.; Botta, F.; Centonze, L.; Najjar, M.; et al. From a Philosophical Framework to a Valid Prognostic Staging System of the New "Comprehensive Assessment" for Transplantable Hepatocellular Carcinoma. Cancers 2019, 11, 741.
- Di Sandro, S.; Benuzzi, L.; Lauterio, A.; Botta, F.; De Carlis, R.; Najjar, M.; Centonze, L.; Danieli, M.; Pezzoli, I.; Rampoldi, A.; et al. Single Hepatocellular Carcinoma approached by curative-intent treatment: A propensity score analysis comparing radiofrequency ablation and liver resection. Eur. J. Surg. Oncol. 2019, 45, 1691–1699.
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63.
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66.
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502.
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173.
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390.
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905.
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296.
- Chen, P.J.; Chen, D.S.; Lai, M.Y.; Chang, M.H.; Huang, G.T.; Yang, P.M.; Sheu, J.C.; Lee, S.C.; Hsu, H.C.; Sung, J.L. Clonal origin of recurrent hepatocellular carcinomas. Gastroenterology 1989, 96, 527–529.
- Chan, L.K.; Tsui, Y.M.; Ho, D.W.; Ng, I.O. Cellular heterogeneity and plasticity in liver cancer. Semin. Cancer Biol. 2021.
- Llovet, J.M.; Schwartz, M.; Mazzaferro, V. Resection and liver transplantation for hepatocellular carcinoma. Semin Liver Dis. 2005, 25, 181–200.
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255.
- Ding, X.; He, M.; Chan, A.W.H.; Song, Q.X.; Sze, S.C.; Chen, H.; Man, M.K.H.; Man, K.; Chan, S.L.; Lai, P.B.S.; et al. Genomic and Epigenomic Features of Primary and Recurrent Hepatocellular Carcinomas. Gastroenterology 2019, 157, 1630–1645.e1636.
- Nault, J.C.; Villanueva, A. Intratumor molecular and phenotypic diversity in hepatocellular carcinoma. Clin. Cancer Res. 2015, 21, 1786–1788.
- Berasain, C.; Avila, M.A. Regulation of hepatocyte identity and quiescence. Cell Mol. Life Sci. 2015, 72, 3831–3851.
- Hernandez-Gea, V.; Toffanin, S.; Friedman, S.L.; Llovet, J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013, 144, 512–527.
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.
- Matson, V.; Chervin, C.S.; Gajewski, T.F. Cancer and the Microbiome-Influence of the Commensal Microbiota on Cancer, Immune Responses, and Immunotherapy. Gastroenterology 2021, 160, 600–613.
- Schwabe, R.F.; Greten, T.F. Gut microbiome in HCC—Mechanisms, diagnosis and therapy. J. Hepatol. 2020, 72, 230–238.
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127.
- Hepatitis viruses. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC Monographs: Lyon, France, 1994; Volume 59, pp. 1–255. ISBN 92832 12592.
- Kew, M.C.; Dusheiko, G.M.; Hadziyannis, S.J.; Patterson, A. Does delta infection play a part in the pathogenesis of hepatitis B virus related hepatocellular carcinoma? Br. Med. J. 1984, 288, 1727.
- Verme, G.; Brunetto, M.R.; Oliveri, F.; Baldi, M.; Forzani, B.; Piantino, P.; Ponzetto, A.; Bonino, F. Role of hepatitis delta virus infection in hepatocellular carcinoma. Dig. Dis. Sci. 1991, 36, 1134–1136.
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1261.
- Zamor, P.J.; de Lemos, A.S.; Russo, M.W. Viral hepatitis and hepatocellular carcinoma: Etiology and management. J. Gastrointest. Oncol. 2017, 8, 229–242.
- Ye, B.; Liu, X.; Li, X.; Kong, H.; Tian, L.; Chen, Y. T-cell exhaustion in chronic hepatitis B infection: Current knowledge and clinical significance. Cell Death Dis. 2015, 6, e1694.
- Heim, M.H.; Thimme, R. Innate and adaptive immune responses in HCV infections. J. Hepatol. 2014, 61, S14–S25.
- Bressac, B.; Kew, M.; Wands, J.; Ozturk, M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 1991, 350, 429–431.
- Bressac, B.; Galvin, K.M.; Liang, T.J.; Isselbacher, K.J.; Wands, J.R.; Ozturk, M. Abnormal structure and expression of p53 gene in human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 1990, 87, 1973–1977.
- Wang, B.; Huang, G.; Wang, D.; Li, A.; Xu, Z.; Dong, R.; Zhang, D.; Zhou, W. Null genotypes of GSTM1 and GSTT1 contribute to hepatocellular carcinoma risk: Evidence from an updated meta-analysis. J. Hepatol. 2010, 53, 508–518.
- Sui, C.; Ma, J.; He, X.; Wang, G.; Ai, F. Interactive effect of glutathione S-transferase M1 and T1 polymorphisms on hepatocellular carcinoma. Tumour Biol. 2014, 35, 8235–8241.
- Schulze, K.; Nault, J.C.; Villanueva, A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J. Hepatol. 2016, 65, 1031–1042.
- Nault, J.C.; De Reyniès, A.; Villanueva, A.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Decaens, T.; Franco, D.; Imbeaud, S.; Rousseau, F.; et al. A hepatocellular carcinoma 5-gene score associated with survival of patients after liver resection. Gastroenterology 2013, 145, 176–187.
- Nault, J.C.; Zucman Rossi, J. Molecular classification of hepatocellular adenomas. Int. J. Hepatol. 2013, 2013, 315947.
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392.
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52.
- Dragani, T.A. Risk of HCC: Genetic heterogeneity and complex genetics. J. Hepatol. 2010, 52, 252–257.
- Bréchot, C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: Old and new paradigms. Gastroenterology 2004, 127, S56–S61.
- Trépo, E.; Nahon, P.; Bontempi, G.; Valenti, L.; Falleti, E.; Nischalke, H.D.; Hamza, S.; Corradini, S.G.; Burza, M.A.; Guyot, E.; et al. Association between the PNPLA3 (rs738409 C>G) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data. Hepatology 2014, 59, 2170–2177.
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465.
- Liu, Y.L.; Patman, G.L.; Leathart, J.B.; Piguet, A.C.; Burt, A.D.; Dufour, J.F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81.
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356.
- Liu, Y.L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.; Allison, M.E.; Alexander, G.J.; Piguet, A.C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309.
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218.
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273.
- Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e1323.
- Lee, J.S.; Heo, J.; Libbrecht, L.; Chu, I.S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 2006, 12, 410–416.
- Kaposi-Novak, P.; Lee, J.S.; Gòmez-Quiroz, L.; Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J. Clin. Investig. 2006, 116, 1582–1595.
- Tovar, V.; Alsinet, C.; Villanueva, A.; Hoshida, Y.; Chiang, D.Y.; Solé, M.; Thung, S.; Moyano, S.; Toffanin, S.; Mínguez, B.; et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J. Hepatol. 2010, 52, 550–559.
- Newell, P.; Toffanin, S.; Villanueva, A.; Chiang, D.Y.; Minguez, B.; Cabellos, L.; Savic, R.; Hoshida, Y.; Lim, K.H.; Melgar-Lesmes, P.; et al. Ras pathway activation in hepatocellular carcinoma and anti-tumoral effect of combined sorafenib and rapamycin in vivo. J. Hepatol. 2009, 51, 725–733.
- Villanueva, A.; Chiang, D.Y.; Newell, P.; Peix, J.; Thung, S.; Alsinet, C.; Tovar, V.; Roayaie, S.; Minguez, B.; Sole, M.; et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008, 135, 1972–1983.
- Villanueva, A.; Alsinet, C.; Yanger, K.; Hoshida, Y.; Zong, Y.; Toffanin, S.; Rodriguez-Carunchio, L.; Solé, M.; Thung, S.; Stanger, B.Z.; et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 2012, 143, 1660–1669.
- Yamashita, T.; Forgues, M.; Wang, W.; Kim, J.W.; Ye, Q.; Jia, H.; Budhu, A.; Zanetti, K.A.; Chen, Y.; Qin, L.X.; et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008, 68, 1451–1461.
- Schlageter, M.; Terracciano, L.M.; D’Angelo, S.; Sorrentino, P. Histopathology of hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 15955–15964.
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761.
- Villanueva, A.; Portela, A.; Sayols, S.; Battiston, C.; Hoshida, Y.; Méndez-González, J.; Imbeaud, S.; Letouzé, E.; Hernandez-Gea, V.; Cornella, H.; et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 2015, 61, 1945–1956.
- Lachenmayer, A.; Alsinet, C.; Savic, R.; Cabellos, L.; Toffanin, S.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Newell, P.; Tsai, H.W.; et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin. Cancer Res. 2012, 18, 4997–5007.
- Chiang, D.Y.; Villanueva, A.; Hoshida, Y.; Peix, J.; Newell, P.; Minguez, B.; LeBlanc, A.C.; Donovan, D.J.; Thung, S.N.; Solé, M.; et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008, 68, 6779–6788.
- Berasain, C.; Ujue Latasa, M.; Urtasun, R.; Goñi, S.; Elizalde, M.; Garcia-Irigoyen, O.; Azcona, M.; Prieto, J.; Avila, M.A. Epidermal Growth Factor Receptor (EGFR) Crosstalks in Liver Cancer. Cancers 2011, 3, 2444–2461.
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892.
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94.
- Dong, A.; Yu, H.; Wang, Y.; Dong, H.; Zuo, C. FDG PET/CT and enhanced CT imaging of tumor heterogeneity in hepatocellular carcinoma: Imaging-pathologic correlation. Clin. Nucl. Med. 2014, 39, 808–810.
- Ishida, Y.; Nagamatsu, H.; Koga, H.; Yoshida, H.; Kojiro, M.; Sata, M. Hepatocellular carcinoma with a “nodule-in-nodule” appearance reflecting an unusual dilated pseudoglandular structure. Intern. Med. 2008, 47, 1215–1218.
- Kim, J.M.; Kwon, C.H.; Joh, J.W.; Park, J.B.; Lee, J.H.; Kim, S.J.; Paik, S.W.; Park, C.K. Intrahepatic metastasis is more risky than multiple occurrence in hepatocellular carcinoma patients after curative liver resection. Hepatogastroenterology 2015, 62, 399–404.
- Yang, S.L.; Luo, Y.Y.; Chen, M.; Zhou, Y.P.; Lu, F.R.; Deng, D.F.; Wu, Y.R. A systematic review and meta-analysis comparing the prognosis of multicentric occurrence and vs. intrahepatic metastasis in patients with recurrent hepatocellular carcinoma after hepatectomy. HPB 2017, 19, 835–842.
- Imamura, H.; Matsuyama, Y.; Tanaka, E.; Ohkubo, T.; Hasegawa, K.; Miyagawa, S.; Sugawara, Y.; Minagawa, M.; Takayama, T.; Kawasaki, S.; et al. Risk factors contributing to early and late phase intrahepatic recurrence of hepatocellular carcinoma after hepatectomy. J. Hepatol. 2003, 38, 200–207.
- Wu, J.C.; Huang, Y.H.; Chau, G.Y.; Su, C.W.; Lai, C.R.; Lee, P.C.; Huo, T.I.; Sheen, I.J.; Lee, S.D.; Lui, W.Y. Risk factors for early and late recurrence in hepatitis B-related hepatocellular carcinoma. J. Hepatol. 2009, 51, 890–897.
- Sun, Y.; Wu, L.; Zhong, Y.; Zhou, K.; Hou, Y.; Wang, Z.; Zhang, Z.; Xie, J.; Wang, C.; Chen, D.; et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 2021, 184, 404–421.
- Torrecilla, S.; Sia, D.; Harrington, A.N.; Zhang, Z.; Cabellos, L.; Cornella, H.; Moeini, A.; Camprecios, G.; Leow, W.Q.; Fiel, M.I.; et al. Trunk mutational events present minimal intra- and inter-tumoral heterogeneity in hepatocellular carcinoma. J. Hepatol. 2017, 67, 1222–1231.
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; von Felden, J.; Labgaa, I.; D’Avola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291.
- Lin, D.C.; Mayakonda, A.; Dinh, H.Q.; Huang, P.; Lin, L.; Liu, X.; Ding, L.W.; Wang, J.; Berman, B.P.; Song, E.W.; et al. Genomic and Epigenomic Heterogeneity of Hepatocellular Carcinoma. Cancer Res. 2017, 77, 2255–2265.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
03 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No