 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Elisa Drago-Serrano | + 5654 word(s) | 5654 | 2021-05-27 10:27:38 | | | |
| 2 | Peter Tang | Meta information modification | 5654 | 2021-06-02 05:00:36 | | |
Video Upload Options
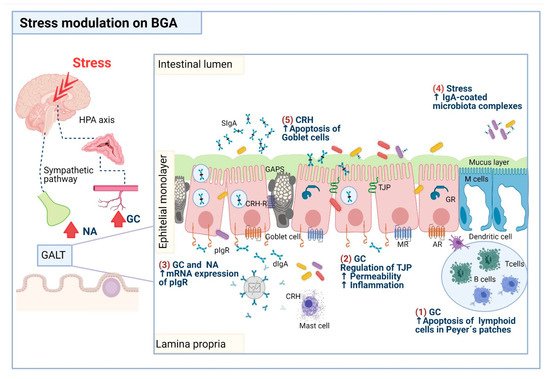
Intestinal homeostasis encompasses a complex and balanced interplay among a wide array of components that collaborate to maintain gut barrier integrity. The appropriate function of the gut barrier requires the mucus layer, a sticky cushion of mucopolysaccharides that overlays the epithelial cell surface. Mucus plays a critical anti-inflammatory role by preventing direct contact between luminal microbiota and the surface of the epithelial cell monolayer. Moreover, mucus is enriched with pivotal effectors of intestinal immunity, such as immunoglobulin A (IgA). A fragile and delicate equilibrium that supports proper barrier function can be disturbed by stress. The impact of stress upon intestinal homeostasis results from neuroendocrine mediators of the brain-gut axis (BGA), which comprises a nervous branch that includes the enteric nervous system (ENS) and the sympathetic and parasympathetic nervous systems, as well as an endocrine branch of the hypothalamic-pituitary-adrenal axis.
1. Introduction
2. An Overview of the Biochemical and Immune Gut Barrier Components
3. Overview of Neuroendocrine Stress Pathways
4. Stress and Immunoglobulin A
|
Chronic Stress Animal Models |
|
|---|---|
|
Model |
Effect |
|
WAS for 1 h a day for 7 days in antibiotic-treated C57BL/6 mice |
↑ IgA cecal content (not significant), ↑ corticosterone, ↑ luminal bacteria adherence, ↑ dysbiosis, ↑ Clostridium spp., ↑ mature goblet cell density, ↓ Verrucobacteria [46]. |
|
Mice exposed to sCSDS 10 days |
↓ IgA cecum; IgA and sCSDS levels were correlated, ↓ mRNA IgA response, ↑ cecal dysbiosis [47]. |
|
Necrotizing enterocolitis-like murine model in offspring of dams that underwent stress |
In offspring from stressed dams: ↓ fecal IgA, ↔ milk IgA. Female offspring of stressed dams: ↑ IgA-bound microbiota, ↑ dysbiosis, ↑ colonic Necrotizing enterocolitis-like injury [48]. |
|
Restraint stress for 1 h a day for 7 days in male Fisher rats prior to MCAO |
↓ IgA colon, ↑ plasma corticosterone, ↑ bacterial translocation to MLN [49]. |
|
Alternating transfer stress in male Sprague Dawley rats (home cage to metabolic cage) |
↓ IgA fecal, ↔ fecal and urine corticosterone [50]. |
|
Maternal separation stress in neonatal rats |
At posnatal day 35 in rats: ↑ intestinal permeability, ↓ intestinal mucin, ↑ dysbiosis [51]. |
|
Restraint stress for 1 h a day for 4 days in male BALB/c mice |
↓ IgA small intestine, ↑ plasma corticosterone and norepinephrine [52]. |
|
Restraint stress for 1 h a day for 4 days in male BALB/c mice |
↓ intraepithelial lymphocytes in the proximal small intestine [53]. |
|
Heat stress for 2 h a day for 3 days in Sprague Dawley rats |
↑ goblet cell gaps in small intestine, ↓ jejunal SIgA, TLR2, TLR4 proteins, ↓ jejunal IL-2, IL-4, IL-10, IFN-γ mRNA, ↑ small intestine injury, ↑ Escherichia coli translocation to MLN [54]. |
|
Chronic restraint stress for 1 h or 4 h a day for 4 days in male BALB/c mice |
↓ IgA+ plasma cells small intestine, ↓ CD8+T and B cells small intestine, ↓ Peyer’s patches cells small intestine [55]. |
|
Restraint stress for 2 h a day for 7 days in C57BL/6J SPF mice |
↑ fecal IgA-bound to bacteria ↑IgA microbiota response, ↑ opening colonic goblet cells associated gaps, ↓ weight loss, diarrhea, ↑ aerobic bacterial translocation to MLN, ↑ dysbiosis [56]. |
|
Restraint for 3 h for 7 days in Wistar rats |
↑ IgA levels, ↑ α-chain mRNA proximal and distal small intestine [57]. |
|
WAS for 1 h or 1 h a day for 5 days for 12 weeks in T cell receptor α chain gene (Tcra−/−) knock out mice |
↑ IgA microbiota response, ↓ microbiota diversity, ↑colitis, ↑dysbiosis in Tcra−/− C57BL/6 mice but not in Tcra−/− BALB/c mice [58]. |
|
WAS for 1 h a day for 10 days in mast-cell-deficient ws/ws rats and wild-type control rats |
↑corticosterone, ↑ macromolecular permeability, ↑ mucus depletion, ↑ mitochondria enlargement and autophagosomes in epithelial cell layer, ↑ bacterial adherence and penetration into enterocytes, neutrophil, and monocyte infiltration, ↑ mieloperoxidase activity, hyperplasia, and activation of mast cells. No changes in ws/ws rats [59]. |
|
Restraint stress for 12 h in male BALB/c mice |
↑ Peyer’s patches apoptosis, ↓ TCD3+ cells and ↓ B220+ cells [60]. |
|
PS or EFS 2 h a day for 14 days in Sprague Dawley rats |
↓ IgA (PS) MLN secretions, ↓ IgG (PS) plasma, ↑ IgA (EFS) MLN secretions, ↑ IgG (EFS) plasma, ↑ corticosterone (EFS) plasma [61]. |
|
Acute stress animal models |
|
|
WIRS 4 h in BALB/c mice that underwent TNBS-ethanol induced ulcerative colitis |
In mice that underwent TNBS-induced ulcerative colitis, stress aggravated: ↓ colonic total and SIgA, ↓ IgA serum, ↑colonic mucosa injury, ↓ goblet cells, ↑ IL-6, -8, TNF-α in serum [62]. |
|
Restraint stress for 6 h male Wistar rats |
↓ colonic IgA, ↔ plasma corticosterone, ↑ bacterial translocation to MLN [63]. |
|
Acute restraint stress for 12 h in mice |
↓ T and B cells, ↑ Peyer’s patches apoptosis, ↑ endogenous glucocorticoids [60]. |
5. Stress and the Mucus Layer
References
- Kayama, H.; Takeda, K. Regulation of Intestinal Homeostasis by Innate and Adaptive Immunity. Int. Immunol. 2012, 24, 673–680.
- Assimakopoulos, S.F.; Triantos, C.; Maroulis, I.; Gogos, C. The Role of the Gut Barrier Function in Health and Disease. Gastroenterol. Res. 2018, 11, 261–263.
- Singhal, R.; Shah, Y.M. Oxygen Battle in the Gut: Hypoxia and Hypoxia-Inducible Factors in Metabolic and Inflammatory Responses in the Intestine. J. Biol. Chem. 2020, 295, 10493–10505.
- Litvak, Y.; Byndloss, M.X.; Bäumler, A.J. Colonocyte Metabolism Shapes the Gut Microbiota. Science 2018, 362.
- Walker, M.Y.; Pratap, S.; Southerland, J.H.; Farmer-Dixon, C.M.; Lakshmyya, K.; Gangula, P.R. Role of Oral and Gut Microbiome in Nitric Oxide-Mediated Colon Motility. Nitric Oxide 2018, 73, 81–88.
- Wells, J.M.; Brummer, R.J.; Derrien, M.; MacDonald, T.T.; Troost, F.; Cani, P.D.; Theodorou, V.; Dekker, J.; Méheust, A.; de Vos, W.M.; et al. Homeostasis of the Gut Barrier and Potential Biomarkers. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G171–G193.
- Kaetzel, C.S.; Mestecky, J.; Johansen, F.-E. Two Cells, One Antibody: The Discovery of the Cellular Origins and Transport of Secretory IgA. J. Immunol. 2017, 198, 1765–1767.
- Bowcutt, R.; Forman, R.; Glymenaki, M.; Carding, S.R.; Else, K.J.; Cruickshank, S.M. Heterogeneity across the Murine Small and Large Intestine. World J. Gastroenterol. 2014, 20, 15216–15232.
- Lu, Z.; Ding, L.; Lu, Q.; Chen, Y.-H. Claudins in Intestines: Distribution and Functional Significance in Health and Diseases. Tissue Barriers 2013, 1, e24978.
- Mowat, A.M.; Agace, W.W. Regional Specialization within the Intestinal Immune System. Nat. Rev. Immunol. 2014, 14, 667–685.
- Brzozowski, B.; Mazur-Bialy, A.; Pajdo, R.; Kwiecien, S.; Bilski, J.; Zwolinska-Wcislo, M.; Mach, T.; Brzozowski, T. Mechanisms by Which Stress Affects the Experimental and Clinical Inflammatory Bowel Disease (IBD): Role of Brain-Gut Axis. Curr. Neuropharmacol. 2016, 14, 892–900.
- Buynitsky, T.; Mostofsky, D.I. Restraint Stress in Biobehavioral Research: Recent Developments. Neurosci. Biobehav. Rev. 2009, 33, 1089–1098.
- Dhabhar, F.S. Enhancing versus Suppressive Effects of Stress on Immune Function: Implications for Immunoprotection and Immunopathology. Neuro Immuno Modul. 2009, 16, 300–317.
- Bhatia, V.; Tandon, R.K. Stress and the Gastrointestinal Tract. J. Gastroenterol. Hepatol. 2005, 20, 332–339.
- Caso, J.; Leza, J.; Menchen, L. The Effects of Physical and Psychological Stress on the Gastrointestinal Tract: Lessons from Animal Models. Curr. Mol. Med. 2008, 8, 299–312.
- De Jonge, W.J. The Gut’s Little Brain in Control of Intestinal Immunity. ISRN Gastroenterol. 2013, 2013.
- Yoo, B.B.; Mazmanian, S.K. The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity 2017, 46, 910–926.
- Lyte, M.; Vulchanova, L.; Brown, D.R. Stress at the Intestinal Surface: Catecholamines and Mucosa-Bacteria Interactions. Cell Tissue Res. 2011, 343, 23–32.
- Oshima, T.; Miwa, H. Gastrointestinal Mucosal Barrier Function and Diseases. J. Gastroenterol. 2016, 51, 768–778.
- Söderholm, J.D.; Perdue, M.H. Stress and the Gastrointestinal Tract II. Stress and Intestinal Barrier Function. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G7–G13.
- Keita, A.V.; Söderholm, J.D. The Intestinal Barrier and Its Regulation by Neuroimmune Factors. Neurogastroenterol. Motil. 2010, 22, 718–733.
- Campos-Rodríguez, R.; Godínez-Victoria, M.; Abarca-Rojano, E.; Pacheco-Yépez, J.; Reyna-Garfias, H.; Barbosa-Cabrera, R.E.; Drago-Serrano, M.E. Stress Modulates Intestinal Secretory Immunoglobulin A. Front. Integr. Neurosci. 2013, 7, 86.
- Costes, L.M.M.; Boeckxstaens, G.E.; de Jonge, W.J.; Cailotto, C. Neural Networks in Intestinal Immunoregulation. Organogenesis 2013, 9, 216–223.
- Konturek, P.C.; Brzozowski, T.; Konturek, S.J. Stress and the Gut: Pathophysiology, Clinical Consequences, Diagnostic Approach and Treatment Options. J. Physiol. Pharmacol. 2011, 62, 591–599.
- Casado-Bedmar, M.; Keita, A.V. Potential Neuro-Immune Therapeutic Targets in Irritable Bowel Syndrome. Ther. Adv. Gastroenterol. 2020, 13, 1756284820910630.
- Friedman, E.S.; Bittinger, K.; Esipova, T.V.; Hou, L.; Chau, L.; Jiang, J.; Mesaros, C.; Lund, P.J.; Liang, X.; FitzGerald, G.A.; et al. Microbes vs. Chemistry in the Origin of the Anaerobic Gut Lumen. Proc. Natl. Acad. Sci. USA 2018, 115, 4170–4175.
- Matheson, P.J.; Wilson, M.A.; Garrison, R.N. Regulation of Intestinal Blood Flow. J. Surg. Res. 2000, 93, 182–196.
- Grondin, J.A.; Kwon, Y.H.; Far, P.M.; Haq, S.; Khan, W.I. Mucins in Intestinal Mucosal Defense and Inflammation: Learning From Clinical and Experimental Studies. Front. Immunol. 2020, 11, 2054.
- Olivares-Villagómez, D.; van Kaer, L. Intestinal Intraepithelial Lymphocytes: Sentinels of the Mucosal Barrier. Trends Immunol. 2018, 39, 264–275.
- Camerini, V.; Panwala, C.; Kronenberg, M. Regional Specialization of the Mucosal Immune System. Intraepithelial Lymphocytes of the Large Intestine Have a Different Phenotype and Function than Those of the Small Intestine. J. Immunol. 1993, 151, 1765–1776.
- Li, Y.; Jin, L.; Chen, T.; Pirozzi, C.J. The Effects of Secretory IgA in the Mucosal Immune System. Biomed. Res. Int. 2020.
- Reboldi, A.; Cyster, J.G. Peyer’s Patches: Organizing B-Cell Responses at the Intestinal Frontier. Immunol. Rev. 2016, 271, 230–245.
- Pabst, O. New Concepts in the Generation and Functions of IgA. Nat. Rev. Immunol. 2012, 12, 821–832.
- MacPherson, A.J.; McCoy, K.D.; Johansen, F.E.; Brandtzaeg, P. The Immune Geography of IgA Induction and Function. Mucosal. Immunol. 2008, 1, 11–22.
- Habtezion, A.; Nguyen, L.P.; Hadeiba, H.; Butcher, E.C. Leukocyte Trafficking to the Small Intestine and Colon. Gastroenterology 2016, 150, 340–354.
- Cerutti, A. The Regulation of IgA Class Switching. Nat. Rev. Immunol. 2008, 8, 421–434.
- Chen, K.; Magri, G.; Grasset, E.K.; Cerutti, A. Rethinking Mucosal Antibody Responses: IgM, IgG and IgD Join IgA. Nat. Rev. Immunol. 2020, 20, 427–441.
- Pietrzak, B.; Tomela, K.; Olejnik-Schmidt, A.; Mackiewicz, A.; Schmidt, M. Secretory Iga in Intestinal Mucosal Secretions as an Adaptive Barrier against Microbial Cells. Int. J. Mol. Sci. 2020, 21, 9254.
- Macpherson, A.J.; Köller, Y.; McCoy, K.D. The Bilateral Responsiveness between Intestinal Microbes and IgA. Trends Immunol 2015, 36, 460–470.
- Pabst, O.; Slack, E. IgA and the Intestinal Microbiota: The Importance of Being Specific. Mucosal. Immunol. 2020, 13, 12–21.
- Liu, Y.; Yuan, X.; Li, L.; Lin, L.; Zuo, X.; Cong, Y.; Li, Y. Increased Ileal Immunoglobulin A Production and Immunoglobulin A-Coated Bacteria in Diarrhea-Predominant Irritable Bowel Syndrome. Clin. Transl. Gastroenterol. 2020, 11, e00146.
- Salerno-Goncalves, R.; Safavie, F.; Fasano, A.; Sztein, M.B. Free and Complexed-Secretory Immunoglobulin A Triggers Distinct Intestinal Epithelial Cell Responses. Clin. Exp. Immunol. 2016, 185, 338–347.
- Stakenborg, N.; di Giovangiulio, M.; Boeckxstaens, G.E.; Matteoli, G. The Versatile Role of the Vagus Nerve in the Gastrointestinal Tract. EMJ Gastroenterol. 2013, 1, 106–114.
- El-Salhy, M.; Danielsson, Å.; Axelsson, H.; Qian, B.F. Neuroendocrine Peptide Levels in the Gastrointestinal Tract of Mice after Unilateral Cervical Vagotomy. Regul. Pept. 2000, 88, 15–20.
- Shibata, M.; Hisajima, T.; Nakano, M.; Goris, R.C.; Funakoshi, K. Morphological Relationships between Peptidergic Nerve Fibers and Immunoglobulin A-Producing Lymphocytes in the Mouse Intestine. Brain Behav. Immun. 2008, 22, 158–166.
- Aguilera, M.; Vergara, P.; Martínez, V. Stress and Antibiotics Alter Luminal and Wall-Adhered Microbiota and Enhance the Local Expression of Visceral Sensory-Related Systems in Mice. Neurogastroenterol. Motil. 2013, 25, e515–e529.
- Aoki-Yoshida, A.; Aoki, R.; Moriya, N.; Goto, T.; Kubota, Y.; Toyoda, A.; Takayama, Y.; Suzuki, C. Omics Studies of the Murine Intestinal Ecosystem Exposed to Subchronic and Mild Social Defeat Stress. J. Proteome Res. 2016, 15, 3126–3138.
- Brawner, K.M.; Yeramilli, V.A.; Kennedy, B.A.; Patel, R.K.; Martin, C.A. Prenatal Stress Increases IgA Coating of Offspring Microbiota and Exacerbates Necrotizing Enterocolitis-like Injury in a Sex-Dependent Manner. Brain Behav. Immun. 2020, 89, 291–299.
- Caso, J.R.; Hurtado, O.; Pereira, M.P.; García-Bueno, B.; Menchén, L.; Alou, L.; Gómez-Lus, M.L.; Moro, M.A.; Lizasoain, I.; Leza, J.C. Colonic Bacterial Translocation as a Possible Factor in Stress-Worsening Experimental Stroke Outcome. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R979–R985.
- Eriksson, E.; Royo, F.; Lyberg, K.; Carlsson, H.E.; Hau, J. Effect of Metabolic Cage Housing on Immunoglobulin A and Corticosterone Excretion in Faeces and Urine of Young Male Rats. Exp. Physiol. 2004, 89, 427–433.
- García-Ródenas, C.L.; Bergonzelli, G.E.; Nutten, S.; Schumann, A.; Cherbut, C.; Turini, M.; Ornstein, K.; Rochat, F.; Corthésy-Theulaz, I. Nutritional Approach to Restore Impaired Intestinal Barrier Function and Growth after Neonatal Stress in Rats. J. Pediatric Gastroenterol. Nutr. 2006, 43, 16–24.
- Jarillo-Luna, A.; Rivera-Aguilar, V.; Garfias, H.R.; Lara-Padilla, E.; Kormanovsky, A.; Campos-Rodríguez, R. Effect of Repeated Restraint Stress on the Levels of Intestinal IgA in Mice. Psychoneuroendocrinology 2007, 32, 681–692.
- Jarillo-Luna, A.; Rivera-Aguilar, V.; Martìnez-Carrillo, B.E.; Barbosa-Cabrera, E.; Garfias, H.R.; Campos-Rodríguez, R. Effect of Restraint Stress on the Population of Intestinal Intraepithelial Lymphocytes in Mice. Brain Behav. Immun. 2008, 22, 265–275.
- Liu, X.; Li, H.; Lu, A.; Zhong, Y.; Hou, X.; Wang, N.; Jia, D.; Zan, J.; Zhao, H.; Xu, J.; et al. Reduction of Intestinal Mucosal Immune Function in Heat-Stressed Rats and Bacterial Translocation. Int. J. Hyperth. 2012, 28, 756–765.
- Martínez-Carrillo, B.E.; Godinez-Victoria, M.; Jarillo-Luna, A.; Oros-Pantoja, R.; Abarca-Rojano, E.; Rivera-Aguilar, V.; Pacheco Yépez, J.; Sánchez-Torres, L.E.; Campos-Rodríguez, R. Repeated Restraint Stress Reduces the Number of IgA-Producing Cells in Peyer’s Patches. NeuroImmunoModulation 2011, 18, 131–141.
- Rengarajan, S.; Knoop, K.A.; Rengarajan, A.; Chai, J.N.; Grajales-Reyes, J.G.; Samineni, V.K.; Russler-Germain, E.V.; Ranganathan, P.; Fasano, A.; Sayuk, G.S.; et al. A Potential Role for Stress-Induced Microbial Alterations in IgA-Associated Irritable Bowel Syndrome with Diarrhea. Cell Rep. Med. 2020, 1, 100124.
- Reyna-Garfias, H.; Miliar, A.; Jarillo-Luna, A.; Rivera-Aguilar, V.; Pacheco-Yepez, J.; Baeza, I.; Campos-Rodríguez, R. Repeated Restraint Stress Increases IgA Concentration in Rat Small Intestine. Brain Behav. Immun. 2010, 24, 110–118.
- Watanabe, Y.; Arase, S.; Nagaoka, N.; Kawai, M.; Matsumoto, S. Chronic Psychological Stress Disrupted the Composition of the Murine Colonic Microbiota and Accelerated a Murine Model of Inflammatory Bowel Disease. PLoS ONE 2016, 11, e0150559.
- Söderholm, J.D.; Yang, P.C.; Ceponis, P.; Vohra, A.; Riddell, R.; Sherman, P.M.; Perdue, M.H. Chronic Stress Induces Mast Cell-Dependent Bacterial Adherence and Initiates Mucosal Inflammation in Rat Intestine. Gastroenterology 2002, 123, 1099–1108.
- Sudo, N.; Oyama, N.; Yu, X.N.; Kubo, C. Restraint Stress-Induced Elevation of Endogenous Glucocorticoids Decreases Peyer’s Patch Cell Numbers via Mechanisms That Are Either Dependent or Independent on Apoptotic Cell Death. NeuroImmunoModulation 2001, 9, 333–339.
- Yamamoto, S.; Motomura, A.; Akahoshi, A.; Takahashi, K.; Minami, H. Immunoglobulin Secretions in the Mesenteric Lymph Node in Stressed Rats. J. Nutr. Sci. Vitaminol. 2009, 55.
- Gong, Y.; Niu, W.; Tang, Y.; Zhang, Q.; Liu, S.; Liu, X.; Wang, X.; Xu, Y. Aggravated Mucosal and Immune Damage in a Mouse Model of Ulcerative Colitis with Stress. Exp. Ther. Med. 2019, 17, 2341–2348.
- Ponferrada, Á.; Caso, J.R.; Alou, L.; Colón, A.; Sevillano, D.; Moro, M.A.; Lizasoain, I.; Menchén, P.; Gómez-Lus, M.L.; Lorenzo, P.; et al. The Role of PPARγ on Restoration of Colonic Homeostasis After Experimental Stress-Induced Inflammation and Dysfunction. Gastroenterology 2007, 132, 1791–1803.
- Shimba, A.; Ikuta, K. Control of Immunity by Glucocorticoids in Health and Disease. Semin. Immunopathol. 2020, 42, 669–680.
- Cima, I.; Corazza, N.; Dick, B.; Fuhrer, A.; Herren, S.; Jakob, S.; Ayuni, E.; Mueller, C.; Brunner, T. Intestinal Epithelial Cells Synthesize Glucocorticoids and Regulate T Cell Activation. J. Exp. Med. 2004, 200, 1635–1646.
- Ince, L.M.; Weber, J.; Scheiermann, C. Control of Leukocyte Trafficking by Stress-Associated Hormones. Front. Immunol. 2019, 9, 3143.
- Li, T.W.; Wang, J.; Lam, J.T.; Gutierrez, E.M.; Solorzano-Vargus, R.S.; Tsai, H.V.; Martín, M.G. Transcriptional Control of the Murine Polymeric IgA Receptor Promoter by Glucocorticoids. Am. J. Physiol. Gastrointest. Liver Physiol. 1999, 276, G1425–G1434.
- Cui, W.; Li, L.X.; Sun, C.M.; Wen, Y.; Zhou, Y.; Dong, Y.L.; Liu, P. Tumor Necrosis Factor Alpha Increases Epithelial Barrier Permeability by Disrupting Tight Junctions in Caco-2 Cells. Braz. J. Med. Biol. Res. 2010, 43, 330–333.
- Lauffer, A.; Vanuytsel, T.; Vanormelingen, C.; Vanheel, H.; Salim Rasoel, S.; Tóth, J.; Tack, J.; Fornari, F.; Farré, R. Subacute Stress and Chronic Stress Interact to Decrease Intestinal Barrier Function in Rats. Stress 2016, 19, 225–234.
- Lee, H.S.; Kim, D.K.; Kim, Y.B.; Lee, K.J. Effect of Acute Stress on Immune Cell Counts and the Expression of Tight Junction Proteins in the Duodenal Mucosa of Rats. Gut Liver 2013, 7, 190–196.
- Zheng, G.; Wu, S.P.; Hu, Y.; Smith, D.E.; Wiley, J.W.; Hong, S. Corticosterone Mediates Stress-Related Increased Intestinal Permeability in a Region-Specific Manner. Neurogastroenterol Motil. 2013, 25, e127–e139.
- Machorro-Rojas, N.; Sainz-Espuñes, T.; Godínez-Victoria, M.; Castañeda-Sánchez, J.I.; Campos-Rodríguez, R.; Pacheco-Yepez, J.; Drago-Serrano, M.E. Impact of Chronic Immobilization Stress on Parameters of Colonic Homeostasis in BALB/c Mice. Mol. Med. Rep. 2019, 20, 2083–2090.
- Cryan, J.F.; O’riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013.
- Matsuo, K.; Zhang, X.; Ono, Y.; Nagatomi, R. Acute Stress-Induced Colonic Tissue HSP70 Expression Requires Commensal Bacterial Components and Intrinsic Glucocorticoid. Brain Behav. Immun. 2009, 23, 108–115.
- Knoop, K.A.; McDonald, K.G.; McCrate, S.; McDole, J.R.; Newberry, R.D. Microbial Sensing by Goblet Cells Controls Immune Surveillance of Luminal Antigens in the Colon. Mucosal. Immunol. 2015, 8, 198–210.
- Rodiño-Janeiro, B.K.; Alonso-Cotoner, C.; Pigrau, M.; Lobo, B.; Vicario, M.; Santos, J. Role of Corticotropin-Releasing Factor in Gastrointestinal Permeability. J. Neurogastroenterol. Motil. 2015, 21, 33–50.
- Castagliuolo, I.; Wershil, B.K.; Karalis, K.; Pasha, A.; Nikulasson, S.T.; Pothoulakis, C. Colonic Mucin Release in Response to Immobilization Stress Is Mast Cell Dependent. Am. J. Physiol. Gastrointest. Liver Physiol. 1998, 274, G1094–G1100.
- Habiyambere, B.; Onyango, E. Chronic Stress Modulates the Mucin Components of the Intestinal Barrier and the Intestinal Morphology. BJMMR 2016, 13, 1–14.
- Estienne, M.; Claustre, J.; Clain-Gardechaux, G.; Paquet, A.; Taché, Y.; Fioramonti, J.; Plaisancié, P. Maternal Deprivation Alters Epithelial Secretory Cell Lineages in Rat Duodenum: Role of CRF-Related Peptides. Gut 2010, 59, 744–751.
- Boudry, G.; Jury, J.; Ping, C.Y.; Perdue, M.H. Chronic Psychological Stress Alters Epithelial Cell Turn-over in Rat Ileum. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1228–G1232.
- Pfeiffer, C.J.; Qiu, B.; Lam, S.K. Reduction of Colonic Mucus by Repeated Short-Term Stress Enhances Experimental Colitis in Rats. J. Physiol. Paris 2001, 95, 81–87.
- Qiu, B.S.; Vallance, B.A.; Blennerhassett, P.A.; Collins, S.M. The Role of CD4+ Lymphocytes in the Susceptibility of Mice to Stress- Induced Reactivation of Experimental Colitis. Nat. Med. 1999, 5, 1178–1182.
- Castagliuolo, I.; LaMont, J.T.; Qiu, B.; Fleming, S.M.; Bhaskar, K.R.; Nikulasson, S.T.; Kornetsky, C.; Pothoulakis, C. Acute Stress Causes Mucin Release from Rat Colon: Role of Corticotropin Releasing Factor and Mast Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 1996, 271, G884–G892.
- Da Silva, S.; Robbe-Masselot, C.; Ait-Belgnaoui, A.; Mancuso, A.; Mercade-Loubière, M.; Salvador-Cartier, C.; Gillet, M.; Ferrier, L.; Loubière, P.; Dague, E.; et al. Stress Disrupts Intestinal Mucus Barrier in Rats via Mucin O-Glycosylation Shift: Prevention by a Probiotic Treatment. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G420–G429.
- Wei, L.; Li, Y.; Tang, W.; Sun, Q.; Chen, L.; Wang, X.; Liu, Q.; Yu, S.; Yu, S.; Liu, C.; et al. Chronic Unpredictable Mild Stress in Rats Induces Colonic Inflammation. Front. Physiol. 2019, 10, 1228.


