 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hong Sheng Cheng | + 3846 word(s) | 3846 | 2021-05-08 04:31:11 | | | |
| 2 | Nora Tang | Meta information modification | 3846 | 2021-05-31 03:13:32 | | | | |
| 3 | Nora Tang | Meta information modification | 3846 | 2021-06-03 05:48:28 | | |
Video Upload Options
Most anticancer therapies target malignant cancer cells while largely ignoring the surrounding noncancer cell components of the tumor or TME. The TME or tumor stroma comprises nonmalignant host cellular and acellular components, including, but not limited to, fibroblasts, immune cells, endothelial cells, fat cells, and noncellular components of the tumor niche such as the basement membrane and ECM. Although most normal host cells in the stroma possess certain tumor-suppressing abilities, the stroma will change during malignancy, causing the tumor stromal cells to confer pro- or anti-tumor properties in a context- and cell type-dependent manner. Over the past decades, the role of the TME in determining every aspect of cancer progression and the efficacy of treatment has become evident. The functions of PPARs in these stromal cells are increasingly appreciated and have direct or indirect impacts on cancer progression.
1. PPARγ: A Master Regulator of Stromal Metabolic Reprogramming
1.1. Cancer-Associated Fibroblasts
Cancer metabolism and bioenergetics are vastly different from those of normal epithelial cells. A high basal metabolic rate, coupled with abnormal vasculatures in the TME, poses a tremendous challenge for cancer cells to fulfill their energy demand. While the cancer cells possess remarkable plasticity and versatility to utilize various substrates to meet their demand for cellular energy, the surrounding stromal cells also play an indispensable role during cancer progression.
Under the paracrine influences of cancer cells, stromal cells such as cancer-associated fibroblasts (CAFs) and cancer-associated adipocytes (CAAs) can transform into substrate donors to provide fuels and building blocks, namely glutamine, L-lactate, fatty acids, and ketone bodies. These metabolites are readily channeled into the Krebs cycle and oxidative phosphorylation of the cancer cells for ATP generation [1][2]. PPARγ governs many processes involved in the metabolic remodeling of stromal cells. Clinically, the expression of PPARγ is significantly upregulated in CAFs of cutaneous skin squamous cell carcinoma and colon adenocarcinoma [3][4]. In one study, immortalized human fibroblasts overexpressing PPARγ were more glycolytic, autophagic, and displayed a senescent phenotype [5]. L-lactate secretion also increased by 70% in PPARγ-overexpressing fibroblasts compared to wild-type counterparts [5]. These PPARγ-induced metabolic features are typical in a tumor-supporting stroma, as evidenced by accelerated tumor xenograft growth of MDA-MD-231 breast cancer cells when co-implanted with transgenic fibroblasts overexpressing PPARγ, but not with wild-type fibroblasts [5].
The hypoxic TME further aggravates the autophagic phenotype in tumor stromal cells, suggesting a modifying role of hypoxia-inducible factor 1α (HIF-1α) in PPARγ-dependent autophagy [5][6]. Furthermore, a study on a genetic defect (MTO1 deficiency) in mitochondria reported that AMP-activated protein kinase (AMPK) and uncoupling protein 2 (UCP2) interacted closely with PPARγ and HIF-1α, generating a HIF1α-PPARγ-UCP2-AMPK axis, to influence mitochondrial bioenergetics and key metabolic processes such as glycolysis, fatty acid oxidation, and oxidative phosphorylation, leading to extensive metabolic reprogramming in fibroblasts [7]. AMPK ensures the maturation of autophagosome and lysosomal fusion during autophagy [8], besides modulating the genes responsible for mitochondrial integrity (UCP2 and PGC-1α), autophagy (BECN-1, LC3B, ATG5, ATG7, and SQSTM1), and mitophagy (PINK1, FUNDC1, BNIP3, and PRKN) [9]. The expression of AMPK target genes is considerably disrupted in fibroblasts overexpressing PPARγ under normoxia and hypoxia [5]. As such, the interplay among PPARγ, HIF1α, and AMPK is pivotal in modulating CAF autophagy, but the exact mode of interaction remains largely elusive.
Following autophagy, glycolysis occurs to recycle cellular organelles and debris into basic building blocks reusable by cancer cells [10][11]. Many glycolytic genes are subject to PPARγ regulation [12][13]. Several studies also pointed to NF-κB as a key transcription factor of stromal autophagy and glycolysis [5][14], but its interaction with PPARγ remains elusive. In short, PPARγ regulates key genes and cellular events in CAFs to accomplish the metabolic coupling of tumor stroma and epithelium, essentially transforming CAFs into a powerhouse that constantly generates energetic biomolecules to support tumor growth.
In contrast to the tumor-supporting properties of CAFs overexpressing PPARγ, pharmacologic PPARγ activation in tumor epithelium confers anticancer effects by reducing tumor proliferation and neovascularization [5]. Thus, the activation of PPARγ metabolically reprograms CAFs to favor autophagic and glycolytic behaviors, allowing cancer cells to use nutrients from non-autonomous sources to sustain their uncontrolled proliferation and other activities.
1.2. Cancer-Associated Adipocytes
Like CAFs, CAAs also serve as storage sites and nutrient donors in the TME [15]. Fibroblasts and mesenchymal stromal cells readily undergo adipogenesis and differentiate into adipocytes upon exposure to adipogenic stimuli, especially the activation and upregulation of PPARγ [16][17]. Cancer exosomes loaded with miRNA-144 and miRNA-155 facilitate the beige/brown differentiation of CAAs by modulating the MAP3K8-Erk1/2-PPARγ axis, whereas those carrying miRNA-126 can disrupt IRS-GLUT4 signaling and promote AMPK- and HIF1α-mediated autophagy [18][19]. Cancer cells can also initiate the dedifferentiation of adjacent adipocytes, a process that is consistently observed when adipocytes are cocultured with cancer cells [20][21]. The process is characterized by the progressive loss of mature adipocyte markers such as leptin, adiponectin, HSL, and PPARγ, increased expression of fibroblast markers such as matrix metalloproteinase 11 (MMP11), collagen I, and α-SMA, as well as the adoption of a fibroblast-like morphology in the cocultured adipocytes [20][21]. These dedifferentiated adipocytes exhibit transcriptional suppression of GLUT4 and IRS1 and inhibit insulin-induced Akt phosphorylation [20]. These aberrations occur alongside the downregulation of MAP3K8-Erk1/2-PPARγ, effectively escalating the catabolic capacity of CAAs to secrete pyruvate, L-lactate, and ketone bodies [18].
Moreover, diminished ligand activation of PPARγ through the constitutive expression of Notch1 induces adipocyte de-differentiation and tumor-like manifestations [22]. Treatment with rosiglitazone, a PPARγ agonist, effectively promoted adipocyte redifferentiation and attenuated the transformation of the adipocytes [22]. Consistent with these observations, the adipocyte-specific deletion of PPARγ in a chemically induced breast cancer model impaired BRCA1 expression in CAAs and subsequently accelerated tumor formation and progression [23]. Undoubtedly, PPARγ is a critical mediator in the cellular fate and metabolic reprogramming of CAAs. Although the actual functionality of adipocyte dedifferentiation in tumor stroma remains unclear, it is generally associated with pro-tumorigenic activities [18][20]. Furthermore, dedifferentiated adipocytes can be redifferentiated into other cell lineages, including beige/brown adipocytes that readily release bioenergetic molecules into the TME [24]. Such plasticity of adipocytes entails the possibility for tumor cells to coerce the CAAs into other tumor supportive cells.
Taken together, CAFs and CAAs are two key stromal cells that undergo extensive metabolic reprogramming to act as energy reserves for cancer epithelium, as illustrated in Figure 1. PPARγ signaling is implicated in the remodeling of both stromal cells, but the activity is vastly different. Autophagic CAFs are triggered by PPARγ activation, while PPARγ is suppressed in dedifferentiated CAAs. This cell type-dependent disparity highlights a need for strategies to target PPARγ in a cell-specific manner so that the treatment is not counter-productive.
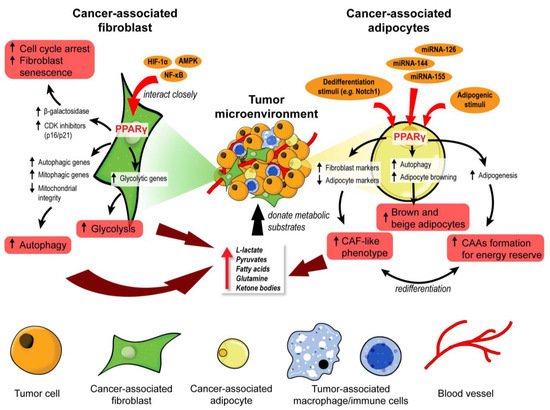
Figure 1. PPARγ orchestrates the metabolic reprogramming of cancer-associated fibroblasts and adipocytes. In cancer-associated fibroblasts (CAFs), PPARγ interacts closely with HIF-1α, AMPK, and NF-κB to promote cell cycle arrest, senescence, autophagy, and glycolysis. These functional changes unleash many metabolic substrates into the tumor microenvironment for the neighboring tumor cells. Similarly, PPARγ governs the fate and function of cancer-associated adipocytes (CAAs). Upon exposure to adipogenic stimuli, PPARγ mediates adipogenesis and formation of CAAs to act as an energy reserve. In contrast, exposure to dedifferentiation stimuli drives CAAs to adopt a CAF-like phenotype and act as a substrate doner in the tumor microenvironment. Certain miRNAs can suppress PPARγ to induce brown and beige differentiation of CAAs which are also energy donors for cancer progression.
2. PPARβ/δ in CAFs Governs Redox Homeostasis and Affects Tumor Initiation
The differentiation of normal fibroblasts into CAFs is one of the cornerstones of early tumor initiation in many cancer types [25][26]. CAFs can disrupt the local ECM and deliver proliferative paracrine signals to support tumorigenic events. Interestingly, mice with fibroblast-selective PPARβ/δ deletion developed fewer and smaller skin tumors than wild-type mice exposed to topical carcinogens [27]. Similar results were recapitulated using chemically and genetically induced intestinal carcinogenesis in these mutant mice [28], indicating that PPARβ/δ activity in stromal fibroblasts promotes tumor initiation. The delayed tumor emergence in the mutant mice was due to an enhanced antioxidant response in the epithelium. Mechanistically, PPARβ/δ-knockout fibroblasts markedly increase the Nox4-derived H2O2 production in the adjacent epidermis, subsequently triggering an RAF/MEK-mediated NRF2 activation that elicits a strong antioxidant and cytoprotective response [27]. By reducing the phosphorylation of many tumor suppressors and oncogenes, NRF2 also increases the tumor suppressor activity of PTEN and reduces the oncogenic activity of Src and Akt, leading to delayed tumor growth [27]. Hence, reducing the expression and activity of PPARβ/δ in CAFs may provide a new therapeutic option to disrupt cancer susceptibility in the neighboring tumor epidermis.
Leucine-rich-alpha-2-glycoprotein 1 (LRG1) and TGFβ1 underpin a crucial process in the PPARβ/δ-mediated stromal–epithelial crosstalk. PPARβ/δ in fibroblasts upregulates the expression of LRG1, which blunts the epidermal response to TGFβ1 [29]. Furthermore, exogenous LRG1 can also ablate the influence of TGFβ1 on ROS generation and NRF2 activity [27]. In colorectal carcinoma and pancreatic ductal adenocarcinoma patients, the level of LRG1 in the TME and bloodstream is significantly higher than in healthy individuals and correlates positively with a more advanced cancer stage and poorer prognosis [30][31][32]. This observation suggests a pro-tumorigenic role of LRG1. Surprisingly, the LRG1 promoter has two putative PPAR response elements [33]. The expression of LRG1 is increased by a PPARβ/δ agonist, GW501516, which strongly suggests that LRG1 is a direct target of PPARβ/δ [33]. Therefore, during the early stage of tumorigenesis, CAF PPARβ/δ may stimulate LRG1 expression, which interferes with TGFβ1-dependent redox homeostasis, to support a sustained oncogenic transformation in the surrounding tumor epithelium.
Collectively, these findings uncover a major role for stromal PPARβ/δ in the epithelial–mesenchymal communication and cellular oxidative response in tumor development (Figure 2). Notably, this novel role of PPARβ/δ was primarily documented, so far, in nonmelanoma skin carcinoma and colorectal cancer models. Thus, further validation in other cancer models is necessary.
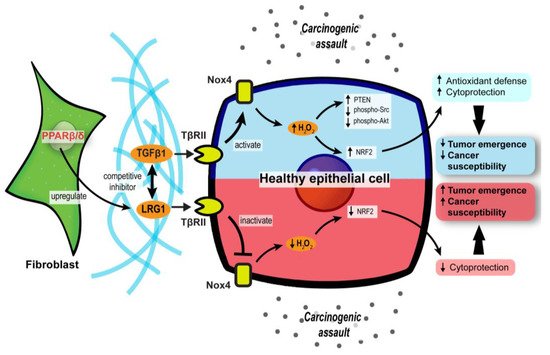
Figure 2. Stromal PPARβ/δ regulates epithelial redox homeostasis and oncogenesis. In carcinogenic assaults, TGFβ signaling in epithelial cells is activated to promote H2O2 synthesis, which subsequently activates NRF2 and reinforces the cytoprotection against carcinogens (blue upper compartment of the epithelial cell). However, fibroblast PPARβ/δ disrupts the protective mechanism by upregulating LRG1, which acts as a competitive inhibitor of TGFβ1 and dampens TGFβ signaling, resulting in increased cancer susceptibility and oncogenesis (red lower compartment of the epithelial cell).
3. Endothelial PPARs Affect Angiogenesis in the Tumor Microenvironment
Hypoxic regions often arise because of rapid tumor growth, which outgrows the oxygen perfusion and nutrient supply from existing vasculature [34]. Cancer cells mitigate the predicament by releasing pro-angiogenic factors that stimulate angiogenesis, which is affected by all three PPAR isotypes.
In terms of PPARα, synthetic PPARα agonists such as fenofibrate and Wy-14643 have demonstrated suppressive effects on endothelial cell proliferation, neovascularization, and tumor xenograft growth [35][36]. Such anti-angiogenic effects of PPARα agonists were lost in PPARα-deficient mice transplanted with PPARα-intact tumor cells, implying that PPARα activation in surrounding stromal cells, but not the tumor cells, attenuated tumor angiogenesis [35][36]. The underlying mechanism is associated with increased anti-angiogenic factors (i.e., thrombospondin-1 and endostatin) and the interference of pro-angiogenic factor biosynthesis (i.e., VEGF-A, angiopoietin-1, and angiopoietin-2), affecting VEGF- and FGF2-mediated endothelial proliferation and migration [35][37]. Furthermore, by transcriptionally suppressing the expression of endothelial P450 CYP2C epoxygenase, whose function is to catalyze arachidonic acid epoxidation, PPARα also diminishes the epoxygenase products, epoxyeicosatrienoic acids, which are pro-angiogenic [38]. Thus, PPARα activation in stromal endothelial cells inhibited the biosynthesis of pro-angiogenic factors while promoting the secretion of anti-angiogenic factors, thereby abrogating angiogenesis and limiting nutrient supply to attenuate tumor progression.
In contrast to PPARα, PPARβ/δ is a pro-angiogenic nuclear receptor in line with its wound healing properties [39][40][41]. The activation of PPARβ/δ in endothelial cells by synthetic ligands or genetic manipulation consistently results in aberrant biosynthesis of VEGF, PDGFR, and c-KI, as well as accelerated endothelial cell proliferation and vascular formation [42][43]. In the TME, these pro-angiogenic changes stimulate the formation of a tumor with a higher vessel density, enhancing tumor feeding, oxygen provision, and metastasis capacity of the cancer cells [43]. Interestingly, in PPARβ/δ knockout mice harboring experimental wild-type tumors, the endothelial cells forming the microvessels in the tumors appear immature, hyperplastic, and less well-organized, leading to abnormal microvasculature and restricted blood flow into the tumors [44][45]. Apart from conventional growth factors, other potential PPARβ/δ-dependent angiogenic mediators include CDKN1C [44], IL-8 [46], CLIC4, and CRBP1 [47]. Considering its regulatory effects on many angiogenic genes and the strong linkages with advanced cancer stages, tumor recurrence, and distant metastasis, PPARβ/δ is identified as one of the pro-angiogenic signaling hubs in cancers [45]. Thus, the pro-tumorigenic and pro-angiogenic activities of PPARβ/δ warrant the development of efficacious PPARβ/δ antagonists to be tested in cancer models.
Existing evidence on the role of PPARγ in angiogenesis remains ambiguous. Like PPARα, PPARγ activities in the TME are associated with the dysregulated production of angiogenic factors, especially platelet-derived endothelial cell growth factor (PD-ECGF) and fibroblast growth factor (FGF) [48][49]. Early studies generally concluded on an inhibitory effect of PPARγ ligands on endothelial cell proliferation in response to pro-angiogenic factors and endothelial tube formation [50][51], whereas subsequent investigations suggested otherwise [52][53]. Such conflicting findings may be attributable to the dosages of PPARγ ligands and endothelial cell types [54]. Regardless of the pro- or anti-angiogenic properties, VEGF/VEGFR signaling is coherently implicated in the PPARγ-mediated effect [50][51][52]. A recent study using endothelial-specific PPARγ knockout models shed new light on the role of this nuclear receptor in angiogenesis. In mature endothelial cells, PPARγ knockdown impaired proliferation, migratory properties, and tubule formation capacity [53]. These impairments translated into the loss of circulating endothelial progenitor cells and angiogenic capacity in endothelial-specific PPARγ-deficient mice, which was reversed by the transplantation of wild-type bone marrow [53]. Mechanistically, abolishing PPARγ in the endothelial cells disrupts E2F1-mediated Wnt signaling and GSK3B interacting protein activity, resulting in suppressed endothelial proliferation [53]. Conceivably, the genetic models reinforce the pro-angiogenic activity of PPARγ in endothelial cells.
In short, PPARα and PPARβ/δ exert anti- and pro-angiogenic activities in the endothelial cells of TME, respectively. On the other hand, opposing roles have been reported for PPARγ in angiogenesis. The roles of each PPAR isotype in angiogenesis are summarized in Figure 3. Notably, most findings on PPARγ are not established using oncogenic models. As the physiological cues in a TME are different from a normal condition, the true nature of PPARγ in cancer angiogenesis and tumor epithelium-endothelium crosstalk requires further investigation.
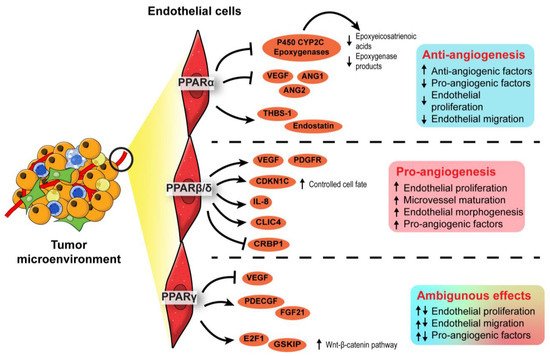
Figure 3. Angiogenic role of PPARs in endothelial cells. In the endothelial cells, PPARα exhibits an anti-angiogenic effect by inhibiting endothelial proliferation, whereas PPARβ/δ appears pro-angiogenic by ensuring proper endothelial morphogenesis and vascular maturation. The role of PPARγ in angiogenesis is conflicting and warrants further investigation.
4. PPAR-Dependent Autocrine and Paracrine Signaling
Autocrine signaling facilities self-stimulation, while paracrine signaling allows local cell–cell communication. In the TME, both forms of cell signaling are imperative to coordinate every stage of oncogenesis, alerting the tumor cells how and when to proliferate, evade immune surveillance, escape from the existing microenvironment, and settle at a distal site. The transmission of complex messages in response to cellular stimuli is made possible by a plethora of secretory mediators, including cytokines, chemokines, growth factors, catalytic proteins, miRNAs, extracellular vesicles, and lipid compounds [55]. Many of these messengers are directly or indirectly regulated by PPARs (Figure 4). For instance, a new PPARγ agonist, CB13, remodels the exosomal contents from radio-resistant non-small cell lung cancer to promote endoplasmic reticulum stress and cell death via a PERK-eIF2α-ATF4-CHOP axis [56].
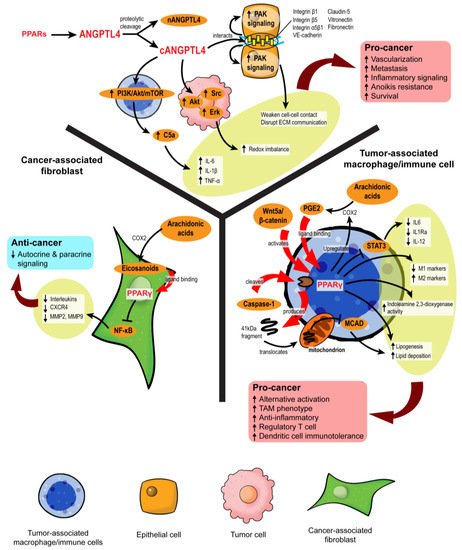
Figure 4. PPARs modulate stromal–epithelial crosstalk in the tumor microenvironment. PPARs affect autocrine and paracrine signaling in different stromal cells. In cancer-associated fibroblasts, PPARγ activation upon ligand binding represses NF-κB, alleviating the secretion of many autocrine and paracrine signals. However, in macrophages and immune cells, PPARγ activation is primarily linked to pro-cancer activities, such as the formation of tumor-associated macrophages (TAMs), increased regulatory T cells, and immunotolerance. ANGPTL4 is a target gene product of PPARs. Proteolytic cleavage of full-length ANGPTL4 yields nANGPTL4 and cANGPTL4 domains, of which the latter is a potent paracrine signal and key mediator of inflammatory signals, anoikis resistance, and metastasis.
4.1. Disruption of Pro-Tumor Signaling by PPARγ in CAFs
Eicosanoids, which are lipid signaling molecules and cognate ligands of PPARs, are the main drivers of PPAR activation in the TME. Major eicosanoid subfamilies include prostaglandins, thromboxanes, leukotrienes, and epoxygenated fatty acids, among which the prostaglandins are the most well-investigated. In colon cancers, cyclooxygenase-2 (COX-2), an enzyme that catalyzes the conversion of arachidonic acid to prostaglandin H2 (PGH2), is overexpressed in CAFs surrounding colon adenocarcinomas, leading to a buildup of intratumoral PGE2 [3][57]. However, the resultant activity of PPARs varies across different stromal cells. For instance, 15d-PGJ2 activates PPARγ and suppresses the proliferation of CAFs and expression of the ECM remodeling enzyme, MMP2 [58]. By inhibiting NF-κB, TZD-activated PPARγ substantially lowers the expression of pro-inflammatory, pro-angiogenic, and pro-metastatic signaling molecules in CAFs, including IL-6, IL-8, CXCR4, MMP2, and MMP9, which further dampens pro-tumor crosstalk in the TME [59][60]. The repression of PPARγ activity also disturbs the quiescent state of hepatic and pancreatic stellate cells, compelling their differentiation into CAFs with highly aggressive phenotypes and inducing desmoplasia in the TME [61][62][63][64]. Despite some conflicting results [65], PPARγ in CAFs can disrupt pro-tumorigenic paracrine signaling by suppressing the liberation of cytokines and chemokines.
4.2. PPARγ Propels the Formation of Tumor-Associated Macrophages
The role of PPARs in innate and adaptive immune cells has been extensively studied. Unlike CAFs, the activation of PPARα and PPARγ in macrophages favors an anti-inflammatory tumor-associated macrophage (TAM) phenotype [66][67]. Classical PPARγ ligands, namely rosiglitazone, N-docosahexaenoyl ethanolamide, and N-docosahexaenoyl serotonin, effectively block paracrine signals from cancer cells to sway the fate of macrophages to adopt alternative activation and reduce their STAT3-mediated pro-inflammatory response [67]. In macrophages challenged with pathogens, WY14643 (PPARα agonist) and 15d-PGJ2 (PPARγ agonist) tip the balance towards the M2 phenotype by enhancing the expression of arginase I, Ym1 (chitinase 3-like 3), mannose receptor, TGF-β and increasing phagocytic capacity while diminishing M1 macrophage biomarkers [68]. PPARγ antagonists and macrophage-specific PPARγ ablation attenuate these effects, clearly outlining the dependency of TAM differentiation on PPARγ [69][70].
Mechanistically, PPARγ agonism promotes lipid retention, lipogenesis, and PGE2 secretion in macrophages. The lipid metabolic changes are partly mediated by the Akt/mTOR pathway [71]. On top of its role as a nuclear receptor and transcription factor, PPARγ is subject to cleavage by caspase-1 to yield a 41 kDa fragment that translocates to mitochondria and inhibits medium-chain acyl-CoA dehydrogenase (MCAD). Such a non-canonical peptide–protein interaction can inhibit fatty acid oxidation, further aggravating lipid droplet accumulation and TAM formation [72]. Likewise, in dendritic cells residing in the TME, PPARγ activation directed by Wnt5a/β-catenin paracrine signaling disrupts fatty acid oxidation and indoleamine 2,3-dioxygenase-1 activity, subsequently leading to the generation of regulatory T cells, immunotolerance, and weakened immunotherapy response [73]. These PPARγ activities create a “friendly” TME for cancer survival, which also coincides with the functional trajectory of macrophage PPARβ/δ [74][75].
Nonetheless, some findings support counterarguments. For example, Cheng et al. (2016) [76] identified macrophage PPARγ as a key tumor suppressor and TAM modulator by abolishing Gpr132 expression. Van Ginderachter et al. (2006) [77] agreed that PPARγ was highly expressed in TAMs, but further stimulation with synthetic and natural ligands could sabotage TAM-induced cytotoxic T lymphocyte suppression to confer an anti-tumor effect. The overexpression of PPARγ in macrophages promotes the upregulation of PTEN, which is encapsulated in exosomes. The uptake of these macrophage-derived exosomes by adjacent cancer cells inhibits Akt, p38 MAPK, and migratory properties [78]. Many eicosanoids are also packaged in these exosomes to achieve paracrine stimulation of PPARγ and augment the inhibitory effect on tumor EMT [78].
Taken together, PPARγ acts as a master immuno-metabolic switch in immune cells that govern their fate and tumor-supporting role. Current consensus depicts that PPARγ exhibits a pro-tumorigenic effect in immune cells by promoting alternative activation, which contradicts its anticancer properties in tumor epithelium and CAFs. On the other hand, the related information on other PPAR isotypes in this aspect is somewhat limited. Interestingly, a recent study unveiled that fatty acid-enriched cancer exosomes markedly activate PPARα in tumor-infiltrating dendritic cells, resulting in mitochondrial overdrive and impaired dendritic cell-mediated CD8+ cytotoxic T-cell priming [79]. These exciting findings strongly suggest an immuno-metabolic regulatory role of PPARα in the TME similar to PPARγ. Such a novel activity of PPARα warrants further investigation.
4.3. Role of ANGPTL4 in Stromal–Epithelial Crosstalk
Growing evidence suggests a role of angiopoietin-like 4 (ANGPTL4) in cancer and stromal-epithelial communication. ANGPTL4 is a secretory protein that belongs to a family of ANGPTL proteins that share high amino acid sequence similarity with the angiopoietin (ANG) family [80][81]. Its expression is regulated by all three PPAR isotypes and PGE2, especially during major metabolic challenges such as starvation and hypoxia [81][82][83]. The native full-length ANGPTL4 can undergo proteolytic cleavage to yield C-terminal (cANGPTL4) and N-terminal (nANGPTL4) chains, each with distinct biological activities [84]. The nANGPTL4 domain is primarily responsible for lipid and glucose metabolism, while the cANGPTL4 domain is closely linked to tumorigenic activities, notably angiogenesis, anoikis resistance, and metastasis [85]. Thus, we will be focusing more on the cANGPTL4 fragment.
High expression of ANGPTL4 has been reported in ovarian, urothelial, and breast tumor biopsies, particularly in the CAAs [86][87][88]. The ANGPTL4 overexpression in CAAs is directed by IL-1β from neighboring TAMs with activated NLRC4 inflammasome and can be exacerbated by tumor hypoxia [89], resulting in cANGPTL4 aggregation in the TME. The cANGPTL4 interacts with integrins β1, β5, α5β1, VE-cadherin, and claudin-5 to induce PAK signaling and weaken cell–cell contacts [90][91]. Moreover, it also disrupts cell–ECM communication through its interaction with vitronectin and fibronectin [92]. The destabilization of cell junctions is then translated to greater intratumoral vascularization and migratory capacity of the malignant cells [93][94][95].
By manipulating redox homeostasis and activating several pro-survival mechanisms such as FAK/Src, PI3K/Akt, Erk signaling, ANGPTL4 markedly sharpens the resilience of tumor cells and confers anoikis resistance [96][97][98]. Our latest report showed that exogenous ANGPTL4 activates macrophages and induces hypercytokinemia via PI3K/Akt-mediated complement component 5a (C5a) activation [99]. This finding indicates a modifying role of ANGPTL4 in TAM functionality and paracrine signaling in the TME. Thus, ANGPTL4 may act as a powerful autocrine and paracrine signaling effector of PPARs that can shape a supportive environment for cancer progression. Further investigations on the therapeutic feasibility of targeting ANGPTL4 are warranted.
5. Stromal PPARγ Modulates Tumor Metastasis
Only a handful of studies have investigated stromal PPAR activities on metastasis, and the results are conflicting. In myeloid-derived suppressor cells (MDSCs), deficiency of lysosomal acid lipase (lal−/−) impaired the production of PPARγ ligands, which led to reduced PPARγ activity, ROS accumulation, and mTOR-mediated tumor metastasis [100]. Following intravenous injection of B16 melanoma cells, increased lung metastases were observed in mice with myeloid-specific PPARγ knockout, further reinforcing the role of MDSCs’ PPARγ in metastasis. Contradictorily, a PPARγ agonist, pioglitazone, has been shown to promote alternative activation of macrophages in the TME [101]. These pro-tumorigenic myeloid cells can synthesize TGFβ1 to promote EMT of surrounding tumor cells [102]. Although the true role of stromal PPARγ in metastasis remains debatable, a recent study showed that astrocytes liberate polyunsaturated fatty acids, which are PPARγ agonists, to promote the extravasation of circulating cancer cells into the brain while PPARγ antagonists can reduce brain metastatic burden in vivo [103]. Astrocyte–cancer cell communication is also mediated by TGF-β2 and ANGPTL4, the latter of which is an effector of PPARs [104]. Hence, PPARγ may serve as a nutritional cue to provoke the invasion of metastatic cells into a nutrient-rich environment. The results also argue for the potential use of PPARγ blockade to treat brain metastasis.
References
- Alam, M.M.; Lal, S.; FitzGerald, K.E.; Zhang, L. A holistic view of cancer bioenergetics: Mitochondrial function and respiration play fundamental roles in the development and progression of diverse tumors. Clin. Transl. Med. 2016, 5, 3.
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60.
- Vandoros, G.P.; Konstantinopoulos, P.A.; Sotiropoulou-Bonikou, G.; Kominea, A.; Papachristou, G.I.; Karamouzis, M.V.; Gkermpesi, M.; Varakis, I.; Papavassiliou, A.G. PPAR-gamma is expressed and NF-κB pathway is activated and correlates positively with COX-2 expression in stromal myofibroblasts surrounding colon adenocarcinomas. J. Cancer Res. Clin. Oncol. 2006, 132, 76–84.
- Chan, J.S.K.; Sng, M.K.; Teo, Z.Q.; Chong, H.C.; Twang, J.S.; Tan, N.S. Targeting nuclear receptors in cancer-associated fibroblasts as concurrent therapy to inhibit development of chemoresistant tumors. Oncogene 2018, 37, 160–173.
- Avena, P.; Anselmo, W.; Whitaker-Menezes, D.; Wang, C.; Pestell, R.G.; Lamb, R.S.; Hulit, J.; Casaburi, I.; Ando, S.; Martinez-Outschoorn, U.E.; et al. Compartment-specific activation of PPARgamma governs breast cancer tumor growth, via metabolic reprogramming and symbiosis. Cell Cycle 2013, 12, 1360–1370.
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284.
- Boutoual, R.; Meseguer, S.; Villarroya, M.; Martin-Hernandez, E.; Errami, M.; Martin, M.A.; Casado, M.; Armengod, M.E. Defects in the mitochondrial-tRNA modification enzymes MTO1 and GTPBP3 promote different metabolic reprogramming through a HIF-PPARgamma-UCP2-AMPK axis. Sci. Rep. 2018, 8, 1163.
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 12637.
- Wang, S.; Kandadi, M.R.; Ren, J. Double knockout of Akt2 and AMPK predisposes cardiac aging without affecting lifespan: Role of autophagy and mitophagy. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 1865–1875.
- Jiao, L.; Zhang, H.L.; Li, D.D.; Yang, K.L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.Y.; Ravichandran, S.; et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy 2018, 14, 671–684.
- Fan, Q.; Yang, L.; Zhang, X.; Ma, Y.; Li, Y.; Dong, L.; Zong, Z.; Hua, X.; Su, D.; Li, H.; et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/beta-catenin signaling pathway activation in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2018, 37, 9.
- Shashni, B.; Sakharkar, K.R.; Nagasaki, Y.; Sakharkar, M.K. Glycolytic enzymes PGK1 and PKM2 as novel transcriptional targets of PPARgamma in breast cancer pathophysiology. J. Drug Target. 2013, 21, 161–174.
- Zhang, H.; Li, L.; Chen, Q.; Li, M.; Feng, J.; Sun, Y.; Zhao, R.; Zhu, Y.; Lv, Y.; Zhu, Z.; et al. PGC1beta regulates multiple myeloma tumor growth through LDHA-mediated glycolytic metabolism. Mol. Oncol. 2018, 12, 1579–1595.
- Londhe, P.; Yu, P.Y.; Ijiri, Y.; Ladner, K.J.; Fenger, J.M.; London, C.; Houghton, P.J.; Guttridge, D.C. Classical NF-kappaB Metabolically Reprograms Sarcoma Cells Through Regulation of Hexokinase 2. Front. Oncol. 2018, 8, 104.
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95.
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Tzekov, A.; Park, C.; Choi, K.; Bickel, P.E. OP9 mouse stromal cells rapidly differentiate into adipocytes: Characterization of a useful new model of adipogenesis. J. Lipid Res. 2006, 47, 450–460.
- Chen, J.H.; Goh, K.J.; Rocha, N.; Groeneveld, M.P.; Minic, M.; Barrett, T.G.; Savage, D.; Semple, R.K. Evaluation of human dermal fibroblasts directly reprogrammed to adipocyte-like cells as a metabolic disease model. Dis. Model. Mech. 2017, 10, 1411–1420.
- Wu, Q.; Li, J.; Li, Z.; Sun, S.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Zhang, Y.; Sun, S.; et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J. Exp. Clin. Cancer Res. 2019, 38, 223.
- Wu, Q.; Sun, S.; Li, Z.; Yang, Q.; Li, B.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Yang, C.; et al. Tumour-originated exosomal miR-155 triggers cancer-associated cachexia to promote tumour progression. Mol. Cancer 2018, 17, 155.
- Cai, Z.; Liang, Y.; Xing, C.; Wang, H.; Hu, P.; Li, J.; Huang, H.; Wang, W.; Jiang, C. Cancer associated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol. Rep. 2019, 42, 2537–2549.
- Zoico, E.; Darra, E.; Rizzatti, V.; Budui, S.; Franceschetti, G.; Mazzali, G.; Rossi, A.P.; Fantin, F.; Menegazzi, M.; Cinti, S.; et al. Adipocytes WNT5a mediated dedifferentiation: A possible target in pancreatic cancer microenvironment. Oncotarget 2016, 7, 20223–20235.
- Bi, P.; Yue, F.; Karki, A.; Castro, B.; Wirbisky, S.E.; Wang, C.; Durkes, A.; Elzey, B.D.; Andrisani, O.M.; Bidwell, C.A.; et al. Notch activation drives adipocyte dedifferentiation and tumorigenic transformation in mice. J. Exp. Med. 2016, 213, 2019–2037.
- Skelhorne-Gross, G.; Reid, A.L.; Apostoli, A.J.; Di Lena, M.A.; Rubino, R.E.; Peterson, N.T.; Schneider, M.; SenGupta, S.K.; Gonzalez, F.J.; Nicol, C.J. Stromal adipocyte PPARgamma protects against breast tumorigenesis. Carcinogenesis 2012, 33, 1412–1420.
- Matsumoto, T.; Kano, K.; Kondo, D.; Fukuda, N.; Iribe, Y.; Tanaka, N.; Matsubara, Y.; Sakuma, T.; Satomi, A.; Otaki, M.; et al. Mature adipocyte-derived dedifferentiated fat cells exhibit multilineage potential. J. Cell Physiol. 2008, 215, 210–222.
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741.
- Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 2256.
- Tan, M.W.Y.; Sng, M.K.; Cheng, H.S.; Low, Z.S.; Leong, B.J.J.; Chua, D.; Tan, E.H.P.; Chan, J.S.K.; Yip, Y.S.; Lee, Y.H.; et al. Deficiency in fibroblast PPARbeta/delta reduces nonmelanoma skin cancers in mice. Cell Death Differ. 2020.
- Tan, E.H.P.; Sng, M.K.; How, I.S.B.; Chan, J.S.K.; Chen, J.; Tan, C.K.; Wahli, W.; Tan, N.S. ROS release by PPARbeta/delta-null fibroblasts reduces tumor load through epithelial antioxidant response. Oncogene 2018, 37, 2067–2078.
- Sng, M.K.; Chan, J.S.K.; Teo, Z.; Phua, T.; Tan, E.H.P.; Wee, J.W.K.; Koh, N.J.N.; Tan, C.K.; Chen, J.P.; Pal, M.; et al. Selective deletion of PPARbeta/delta in fibroblasts causes dermal fibrosis by attenuated LRG1 expression. Cell Discov. 2018, 4, 15.
- Zhang, Q.; Huang, R.; Tang, Q.; Yu, Y.; Huang, Q.; Chen, Y.; Wang, G.; Wang, X. Leucine-rich alpha-2-glycoprotein-1 is up-regulated in colorectal cancer and is a tumor promoter. Onco Targets Ther. 2018, 11, 2745–2752.
- Xie, Z.B.; Zhang, Y.F.; Jin, C.; Mao, Y.S.; Fu, D.L. LRG-1 promotes pancreatic cancer growth and metastasis via modulation of the EGFR/p38 signaling. J. Exp. Clin. Cancer Res. 2019, 38, 75.
- Zhou, Y.; Zhang, X.; Zhang, J.; Fang, J.; Ge, Z.; Li, X. LRG1 promotes proliferation and inhibits apoptosis in colorectal cancer cells via RUNX1 activation. PLoS ONE 2017, 12, e0175122.
- Liu, C.; Lim, S.T.; Teo, M.H.Y.; Tan, M.S.Y.; Kulkarni, M.D.; Qiu, B.; Li, A.; Lal, S.; Dos Remedios, C.G.; Tan, N.S.; et al. Collaborative Regulation of LRG1 by TGF-beta1 and PPAR-beta/delta Modulates Chronic Pressure Overload-Induced Cardiac Fibrosis. Circ. Heart Fail. 2019, 12, e005962.
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190.
- Panigrahy, D.; Kaipainen, A.; Huang, S.; Butterfield, C.E.; Barnes, C.M.; Fannon, M.; Laforme, A.M.; Chaponis, D.M.; Folkman, J.; Kieran, M.W. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 985–990.
- Pozzi, A.; Ibanez, M.R.; Gatica, A.E.; Yang, S.; Wei, S.; Mei, S.; Falck, J.R.; Capdevila, J.H. Peroxisomal proliferator-activated receptor-alpha-dependent inhibition of endothelial cell proliferation and tumorigenesis. J. Biol. Chem. 2007, 282, 17685–17695.
- Arima, T.; Uchiyama, M.; Nakano, Y.; Nagasaka, S.; Kang, D.; Shimizu, A.; Takahashi, H. Peroxisome proliferator-activated receptor alpha agonist suppresses neovascularization by reducing both vascular endothelial growth factor and angiopoietin-2 in corneal alkali burn. Sci. Rep. 2017, 7, 17763.
- Pozzi, A.; Popescu, V.; Yang, S.; Mei, S.; Shi, M.; Puolitaival, S.M.; Caprioli, R.M.; Capdevila, J.H. The anti-tumorigenic properties of peroxisomal proliferator-activated receptor alpha are arachidonic acid epoxygenase-mediated. J. Biol. Chem. 2010, 285, 12840–12850.
- Leu, J.-G.; Chiang, M.-H.; Chen, C.-Y.; Lin, J.-T.; Chen, H.-M.; Chen, Y.-L.; Liang, Y.-J. Adenine accelerated the diabetic wound healing by PPAR delta and angiogenic regulation. Eur. J. Pharmacol. 2018, 818, 569–577.
- Montagner, A.; Wahli, W.; Tan, N.S. Nuclear receptor peroxisome proliferator activated receptor (PPAR) beta/delta in skin wound healing and cancer. Eur. J. Dermatol. 2015, 25 (Suppl. 1), 4–11.
- Tan, N.S.; Icre, G.; Montagner, A.; Bordier-ten-Heggeler, B.; Wahli, W.; Michalik, L. The nuclear hormone receptor peroxisome proliferator-activated receptor beta/delta potentiates cell chemotactism, polarization, and migration. Mol. Cell Biol. 2007, 27, 7161–7175.
- Piqueras, L.; Reynolds, A.R.; Hodivala-Dilke, K.M.; Alfranca, A.; Redondo, J.M.; Hatae, T.; Tanabe, T.; Warner, T.D.; Bishop-Bailey, D. Activation of PPARbeta/delta induces endothelial cell proliferation and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 63–69.
- Wagner, K.D.; Du, S.; Martin, L.; Leccia, N.; Michiels, J.F.; Wagner, N. Vascular PPARbeta/delta Promotes Tumor Angiogenesis and Progression. Cells 2019, 8, 1623.
- Muller-Brusselbach, S.; Komhoff, M.; Rieck, M.; Meissner, W.; Kaddatz, K.; Adamkiewicz, J.; Keil, B.; Klose, K.J.; Moll, R.; Burdick, A.D.; et al. Deregulation of tumor angiogenesis and blockade of tumor growth in PPARbeta-deficient mice. EMBO J. 2007, 26, 3686–3698.
- Abdollahi, A.; Schwager, C.; Kleeff, J.; Esposito, I.; Domhan, S.; Peschke, P.; Hauser, K.; Hahnfeldt, P.; Hlatky, L.; Debus, J.; et al. Transcriptional network governing the angiogenic switch in human pancreatic cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12890–12895.
- Meissner, M.; Hrgovic, I.; Doll, M.; Naidenow, J.; Reichenbach, G.; Hailemariam-Jahn, T.; Michailidou, D.; Gille, J.; Kaufmann, R. Peroxisome proliferator-activated receptor δ activators induce IL-8 expression in nonstimulated endothelial cells in a transcriptional and posttranscriptional manner. J. Biol. Chem. 2010, 285, 33797–33804.
- Adamkiewicz, J.; Kaddatz, K.; Rieck, M.; Wilke, B.; Muller-Brusselbach, S.; Muller, R. Proteomic profile of mouse fibroblasts with a targeted disruption of the peroxisome proliferator activated receptor-beta/delta gene. Proteomics 2007, 7, 1208–1216.
- Possati, L.; Rocchetti, R.; Talevi, S.; Beatrici, V.; Margiotta, C.; Ferrante, L.; Calza, R.; Sagrini, D.; Ferri, A. The role of peroxisome proliferator-activated receptor gamma in bladder cancer in relation to angiogenesis and progression. Gen. Pharmacol. 2000, 35, 269–275.
- Huang, W.; Shao, M.; Liu, H.; Chen, J.; Hu, J.; Zhu, L.; Liu, F.; Wang, D.; Zou, Y.; Xiong, Y.; et al. Fibroblast growth factor 21 enhances angiogenesis and wound healing of human brain microvascular endothelial cells by activating PPARgamma. J. Pharmacol. Sci. 2019, 140, 120–127.
- Xin, X.; Yang, S.; Kowalski, J.; Gerritsen, M.E. Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo. J. Biol. Chem. 1999, 274, 9116–9121.
- Sarayba, M.A.; Li, L.; Tungsiripat, T.; Liu, N.H.; Sweet, P.M.; Patel, A.J.; Osann, K.E.; Chittiboyina, A.; Benson, S.C.; Pershadsingh, H.A.; et al. Inhibition of corneal neovascularization by a peroxisome proliferator-activated receptor-gamma ligand. Exp. Eye Res. 2005, 80, 435–442.
- Biscetti, F.; Gaetani, E.; Flex, A.; Aprahamian, T.; Hopkins, T.; Straface, G.; Pecorini, G.; Stigliano, E.; Smith, R.C.; Angelini, F.; et al. Selective activation of peroxisome proliferator-activated receptor (PPAR)alpha and PPAR gamma induces neoangiogenesis through a vascular endothelial growth factor-dependent mechanism. Diabetes 2008, 57, 1394–1404.
- Vattulainen-Collanus, S.; Akinrinade, O.; Li, M.; Koskenvuo, M.; Li, C.G.; Rao, S.P.; de Jesus Perez, V.; Yuan, K.; Sawada, H.; Koskenvuo, J.W.; et al. Loss of PPARgamma in endothelial cells leads to impaired angiogenesis. J. Cell Sci. 2016, 129, 693–705.
- Bishop-Bailey, D.; Swales, K.E. The Role of PPARs in the Endothelium: Implications for Cancer Therapy. PPAR Res. 2008, 2008, 904251.
- Kusuma, G.D.; Carthew, J.; Lim, R.; Frith, J.E. Effect of the Microenvironment on Mesenchymal Stem Cell Paracrine Signaling: Opportunities to Engineer the Therapeutic Effect. Stem. Cells Dev. 2017, 26, 617–631.
- Kim, T.W.; Hong, D.W.; Hong, S.H. CB13, a novel PPARgamma ligand, overcomes radio-resistance via ROS generation and ER stress in human non-small cell lung cancer. Cell Death Dis. 2020, 11, 848.
- Rigas, B.; Goldman, I.S.; Levine, L. Altered eicosanoid levels in human colon cancer. J. Lab. Clin. Med. 1993, 122, 518–523.
- Paulitschke, V.; Gruber, S.; Hofstatter, E.; Haudek-Prinz, V.; Klepeisz, P.; Schicher, N.; Jonak, C.; Petzelbauer, P.; Pehamberger, H.; Gerner, C.; et al. Proteome analysis identified the PPARgamma ligand 15d-PGJ2 as a novel drug inhibiting melanoma progression and interfering with tumor-stroma interaction. PLoS ONE 2012, 7, e46103.
- Rovito, D.; Gionfriddo, G.; Barone, I.; Giordano, C.; Grande, F.; De Amicis, F.; Lanzino, M.; Catalano, S.; Ando, S.; Bonofiglio, D. Ligand-activated PPARgamma downregulates CXCR4 gene expression through a novel identified PPAR response element and inhibits breast cancer progression. Oncotarget 2016, 7, 65109–65124.
- Papi, A.; De Carolis, S.; Bertoni, S.; Storci, G.; Sceberras, V.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; Orlandi, M.; Bonafe, M. PPARgamma and RXR ligands disrupt the inflammatory cross-talk in the hypoxic breast cancer stem cells niche. J. Cell. Physiol. 2014, 229, 1595–1606.
- Kim, N.; Choi, S.; Lim, C.; Lee, H.; Oh, J. Albumin mediates PPAR-gamma or C/EBP-alpha-induced phenotypic changes in pancreatic stellate cells. Biochem. Biophys. Res. Commun. 2010, 391, 640–644.
- Sharvit, E.; Abramovitch, S.; Reif, S.; Bruck, R. Amplified inhibition of stellate cell activation pathways by PPAR-gamma, RAR and RXR agonists. PLoS ONE 2013, 8, e76541.
- Zhang, Q.; Xiang, S.; Liu, Q.; Gu, T.; Yao, Y.; Lu, X. PPARgamma Antagonizes Hypoxia-Induced Activation of Hepatic Stellate Cell through Cross Mediating PI3K/AKT and cGMP/PKG Signaling. PPAR Res. 2018, 2018, 6970407.
- Shimizu, K.; Kobayashi, M.; Tahara, J.; Shiratori, K. Cytokines and peroxisome proliferator-activated receptor gamma ligand regulate phagocytosis by pancreatic stellate cells. Gastroenterology 2005, 128, 2105–2118.
- Pich, C.; Meylan, P.; Mastelic-Gavillet, B.; Nguyen, T.N.; Loyon, R.; Trang, B.K.; Moser, H.; Moret, C.; Goepfert, C.; Hafner, J.; et al. Induction of Paracrine Signaling in Metastatic Melanoma Cells by PPARgamma Agonist Rosiglitazone Activates Stromal Cells and Enhances Tumor Growth. Cancer Res. 2018, 78, 6447–6461.
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338.
- Gionfriddo, G.; Plastina, P.; Augimeri, G.; Catalano, S.; Giordano, C.; Barone, I.; Morelli, C.; Giordano, F.; Gelsomino, L.; Sisci, D.; et al. Modulating Tumor-Associated Macrophage Polarization by Synthetic and Natural PPARgamma Ligands as a Potential Target in Breast Cancer. Cells 2020, 9, 174.
- Penas, F.; Mirkin, G.A.; Vera, M.; Cevey, A.; Gonzalez, C.D.; Gomez, M.I.; Sales, M.E.; Goren, N.B. Treatment in vitro with PPARalpha and PPARgamma ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim. Biophys. Acta 2015, 1852, 893–904.
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120.
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24.
- Souza-Moreira, L.; Soares, V.C.; Dias, S.; Bozza, P.T. Adipose-derived Mesenchymal Stromal Cells Modulate Lipid Metabolism and Lipid Droplet Biogenesis via AKT/mTOR -PPARgamma Signalling in Macrophages. Sci. Rep. 2019, 9, 20304.
- Niu, Z.; Shi, Q.; Zhang, W.; Shu, Y.; Yang, N.; Chen, B.; Wang, Q.; Zhao, X.; Chen, J.; Cheng, N.; et al. Caspase-1 cleaves PPARgamma for potentiating the pro-tumor action of TAMs. Nat. Commun. 2017, 8, 766.
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-beta-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147–160 e147.
- Schumann, T.; Adhikary, T.; Wortmann, A.; Finkernagel, F.; Lieber, S.; Schnitzer, E.; Legrand, N.; Schober, Y.; Nockher, W.A.; Toth, P.M.; et al. Deregulation of PPARbeta/delta target genes in tumor-associated macrophages by fatty acid ligands in the ovarian cancer microenvironment. Oncotarget 2015, 6, 13416–13433.
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507.
- Cheng, W.Y.; Huynh, H.; Chen, P.; Pena-Llopis, S.; Wan, Y. Macrophage PPARgamma inhibits Gpr132 to mediate the anti-tumor effects of rosiglitazone. Elife 2016, 5, e18502.
- Van Ginderachter, J.A.; Meerschaut, S.; Liu, Y.; Brys, L.; De Groeve, K.; Hassanzadeh Ghassabeh, G.; Raes, G.; De Baetselier, P. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood 2006, 108, 525–535.
- Kim, Y.B.; Ahn, Y.H.; Jung, J.H.; Lee, Y.J.; Lee, J.H.; Kang, J.L. Programming of macrophages by UV-irradiated apoptotic cancer cells inhibits cancer progression and lung metastasis. Cell. Mol. Immunol. 2019, 16, 851–867.
- Yin, X.; Zeng, W.; Wu, B.; Wang, L.; Wang, Z.; Tian, H.; Wang, L.; Jiang, Y.; Clay, R.; Wei, X.; et al. PPARalpha Inhibition Overcomes Tumor-Derived Exosomal Lipid-Induced Dendritic Cell Dysfunction. Cell Rep. 2020, 33, 108278.
- Tan, M.J.; Teo, Z.; Sng, M.K.; Zhu, P.; Tan, N.S. Emerging roles of angiopoietin-like 4 in human cancer. Mol. Cancer Res. 2012, 10, 677–688.
- Zhu, P.; Goh, Y.Y.; Chin, H.F.; Kersten, S.; Tan, N.S. Angiopoietin-like 4: A decade of research. Biosci. Rep. 2012, 32, 211–219.
- Kim, S.H.; Park, Y.Y.; Kim, S.W.; Lee, J.S.; Wang, D.; DuBois, R.N. ANGPTL4 induction by prostaglandin E2 under hypoxic conditions promotes colorectal cancer progression. Cancer Res. 2011, 71, 7010–7020.
- Kersten, S.; Mandard, S.; Tan, N.S.; Escher, P.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J. Biol. Chem. 2000, 275, 28488–28493.
- Ge, H.; Yang, G.; Huang, L.; Motola, D.L.; Pourbahrami, T.; Li, C. Oligomerization and regulated proteolytic processing of angiopoietin-like protein 4. J. Biol. Chem. 2004, 279, 2038–2045.
- La Paglia, L.; Listi, A.; Caruso, S.; Amodeo, V.; Passiglia, F.; Bazan, V.; Fanale, D. Potential Role of ANGPTL4 in the Cross Talk between Metabolism and Cancer through PPAR Signaling Pathway. PPAR Res. 2017, 2017, 8187235.
- Zhou, S.; Wang, R.; Xiao, H. Adipocytes induce the resistance of ovarian cancer to carboplatin through ANGPTL4. Oncol. Rep. 2020, 44, 927–938.
- Cai, Y.C.; Yang, H.; Wang, K.F.; Chen, T.H.; Jiang, W.Q.; Shi, Y.X. ANGPTL4 overexpression inhibits tumor cell adhesion and migration and predicts favorable prognosis of triple-negative breast cancer. BMC Cancer 2020, 20, 878.
- Hsieh, H.Y.; Jou, Y.C.; Tung, C.L.; Tsai, Y.S.; Wang, Y.H.; Chi, C.L.; Lin, R.I.; Hung, S.K.; Chuang, Y.M.; Wu, S.F.; et al. Epigenetic silencing of the dual-role signal mediator, ANGPTL4 in tumor tissues and its overexpression in the urothelial carcinoma microenvironment. Oncogene 2018, 37, 673–686.
- Kolb, R.; Kluz, P.; Tan, Z.W.; Borcherding, N.; Bormann, N.; Vishwakarma, A.; Balcziak, L.; Zhu, P.; Davies, B.S.; Gourronc, F.; et al. Obesity-associated inflammation promotes angiogenesis and breast cancer via angiopoietin-like 4. Oncogene 2019, 38, 2351–2363.
- Goh, Y.Y.; Pal, M.; Chong, H.C.; Zhu, P.; Tan, M.J.; Punugu, L.; Lam, C.R.; Yau, Y.H.; Tan, C.K.; Huang, R.L.; et al. Angiopoietin-like 4 interacts with integrins beta1 and beta5 to modulate keratinocyte migration. Am. J. Pathol. 2010, 177, 2791–2803.
- Huang, R.L.; Teo, Z.; Chong, H.C.; Zhu, P.; Tan, M.J.; Tan, C.K.; Lam, C.R.; Sng, M.K.; Leong, D.T.; Tan, S.M.; et al. ANGPTL4 modulates vascular junction integrity by integrin signaling and disruption of intercellular VE-cadherin and claudin-5 clusters. Blood 2011, 118, 3990–4002.
- Goh, Y.Y.; Pal, M.; Chong, H.C.; Zhu, P.; Tan, M.J.; Punugu, L.; Tan, C.K.; Huang, R.L.; Sze, S.K.; Tang, M.B.; et al. Angiopoietin-like 4 interacts with matrix proteins to modulate wound healing. J. Biol. Chem. 2010, 285, 32999–33009.
- Nakayama, T.; Hirakawa, H.; Shibata, K.; Nazneen, A.; Abe, K.; Nagayasu, T.; Taguchi, T. Expression of angiopoietin-like 4 (ANGPTL4) in human colorectal cancer: ANGPTL4 promotes venous invasion and distant metastasis. Oncol. Rep. 2011, 25, 929–935.
- Ma, T.; Jham, B.C.; Hu, J.; Friedman, E.R.; Basile, J.R.; Molinolo, A.; Sodhi, A.; Montaner, S. Viral G protein-coupled receptor up-regulates Angiopoietin-like 4 promoting angiogenesis and vascular permeability in Kaposi’s sarcoma. Proc. Natl. Acad. Sci. USA 2010, 107, 14363–14368.
- Huang, X.F.; Han, J.; Hu, X.T.; He, C. Mechanisms involved in biological behavior changes associated with Angptl4 expression in colon cancer cell lines. Oncol. Rep. 2012, 27, 1541–1547.
- Zhu, P.; Tan, M.J.; Huang, R.L.; Tan, C.K.; Chong, H.C.; Pal, M.; Lam, C.R.; Boukamp, P.; Pan, J.Y.; Tan, S.H.; et al. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2(-):H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell 2011, 19, 401–415.
- Baba, K.; Kitajima, Y.; Miyake, S.; Nakamura, J.; Wakiyama, K.; Sato, H.; Okuyama, K.; Kitagawa, H.; Tanaka, T.; Hiraki, M.; et al. Hypoxia-induced ANGPTL4 sustains tumour growth and anoikis resistance through different mechanisms in scirrhous gastric cancer cell lines. Sci. Rep. 2017, 7, 11127.
- Liao, Y.H.; Chiang, K.H.; Shieh, J.M.; Huang, C.R.; Shen, C.J.; Huang, W.C.; Chen, B.K. Epidermal growth factor-induced ANGPTL4 enhances anoikis resistance and tumour metastasis in head and neck squamous cell carcinoma. Oncogene 2017, 36, 2228–2242.
- Jung, K.H.; Son, M.K.; Yan, H.H.; Fang, Z.; Kim, J.; Kim, S.J.; Park, J.H.; Lee, J.E.; Yoon, Y.C.; Seo, M.S.; et al. ANGPTL4 exacerbates pancreatitis by augmenting acinar cell injury through upregulation of C5a. EMBO Mol. Med. 2020, 12, e11222.
- Zhao, T.; Du, H.; Blum, J.S.; Yan, C. Critical role of PPARgamma in myeloid-derived suppressor cell-stimulated cancer cell proliferation and metastasis. Oncotarget 2016, 7, 1529–1543.
- Li, H.; Sorenson, A.L.; Poczobutt, J.; Amin, J.; Joyal, T.; Sullivan, T.; Crossno, J.T., Jr.; Weiser-Evans, M.C.; Nemenoff, R.A. Activation of PPARgamma in myeloid cells promotes lung cancer progression and metastasis. PLoS ONE 2011, 6, e28133.
- Sippel, T.R.; Johnson, A.M.; Li, H.Y.; Hanson, D.; Nguyen, T.T.; Bullock, B.L.; Poczobutt, J.M.; Kwak, J.W.; Kleczko, E.K.; Weiser-Evans, M.C.; et al. Activation of PPARgamma in Myeloid Cells Promotes Progression of Epithelial Lung Tumors through TGFbeta1. Mol. Cancer Res. 2019, 17, 1748–1758.
- Zou, Y.; Watters, A.; Cheng, N.; Perry, C.E.; Xu, K.; Alicea, G.M.; Parris, J.L.D.; Baraban, E.; Ray, P.; Nayak, A.; et al. Polyunsaturated Fatty Acids from Astrocytes Activate PPARgamma Signaling in Cancer Cells to Promote Brain Metastasis. Cancer Discov. 2019, 9, 1720–1735.
- Gong, X.; Hou, Z.; Endsley, M.P.; Gronseth, E.I.; Rarick, K.R.; Jorns, J.M.; Yang, Q.; Du, Z.; Yan, K.; Bordas, M.L.; et al. Interaction of tumor cells and astrocytes promotes breast cancer brain metastases through TGF-β2/ANGPTL4 axes. NPJ Precis. Oncol. 2019, 3, 24.


