1000/1000
Hot
Most Recent
 +1 point
+1 point
Mitophagy is a selective autophagic process that eliminates unnecessary and/or damaged mitochondria. Therefore, it is a central hormetic mechanism of mitochondrial quality and quantity control, essential for cellular homeostasis. Its dysregulation has been shown to be a key event in metabolic related diseases and it is the target of emerging therapeutical approaches in this field.
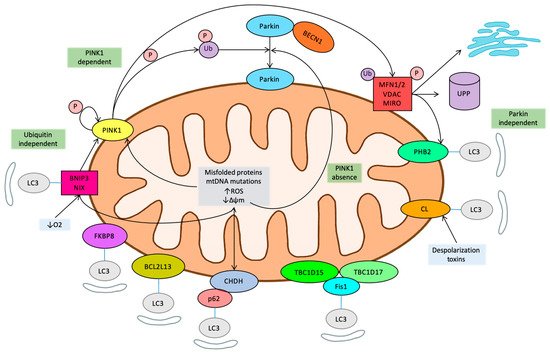
The central relevance of mitophagy in all diseases related to metabolic control is now well established. As mitophagy is required to control metabolic homeostasis or remove damaged or unnecessary mitochondria, it prevents mitochondrial malfunction and subsequent molecular events such as oxidative stress that lead to disease development. In diseased states, mitophagy can sometimes partially compensate other deficits alleviating them, but when mitochondrial activity is compromised, mitophagy can actually play a detrimental role. This is specially evidenced in diseases where normal mitophagy activity is compromised by genetic or regulatory events. These results have boosted pharmacological research in the field, since there are several potentially druggable targets along the mitophagy pathway. Some of these molecules are already showing promising results and more will certainly come soon. Several natural dietary compounds, such as polyphenols, flavonoids, spermidine, or trehalose, which restore normal mitophagy fluxes in the elderly [82][83], could help to control the inflammasome and to prevent neurodegeneration having a direct impact on apoptosis and the caspase activation cascade [84][85]. Perhaps more importantly, lifestyle interventions can promote cardiovascular health, boosting mitophagy [86][87]. The future could also bring new findings on novel, non-canonical mechanisms of mitochondrial quality control, such as the recently described mitochondrion-derived vesicles, characterized in hypoxic neurons and cardiomyocytes [88].





