Mitophagy is a selective autophagic process that eliminates unnecessary and/or damaged mitochondria. Therefore, it is a central hormetic mechanism of mitochondrial quality and quantity control, essential for cellular homeostasis. Its dysregulation has been shown to be a key event in metabolic related diseases and it is the target of emerging therapeutical approaches in this field.
- Mitophagy
- parkin
- PINK1
- autophagy
- human diseases
1. History
2. Mitophagy, a Type of Autophagy
3. PINK1
4. Mitochondrial Homeostasis-Related Pathways
5. LC3
6. Ubiquitin Independent Mitophagy
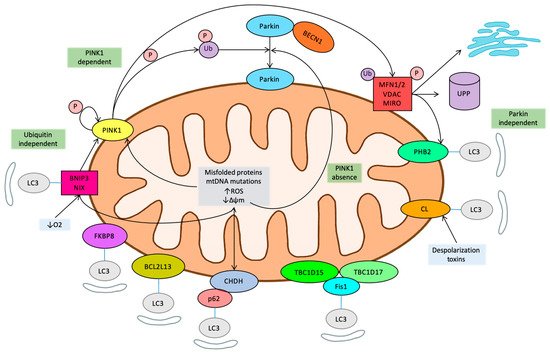
7. Novel Regulatory Pathways
8. Implications for human diseases and therapeutical approaches
The central relevance of mitophagy in all diseases related to metabolic control is now well established. As mitophagy is required to control metabolic homeostasis or remove damaged or unnecessary mitochondria, it prevents mitochondrial malfunction and subsequent molecular events such as oxidative stress that lead to disease development. In diseased states, mitophagy can sometimes partially compensate other deficits alleviating them, but when mitochondrial activity is compromised, mitophagy can actually play a detrimental role. This is specially evidenced in diseases where normal mitophagy activity is compromised by genetic or regulatory events. These results have boosted pharmacological research in the field, since there are several potentially druggable targets along the mitophagy pathway. Some of these molecules are already showing promising results and more will certainly come soon. Several natural dietary compounds, such as polyphenols, flavonoids, spermidine, or trehalose, which restore normal mitophagy fluxes in the elderly [82][83], could help to control the inflammasome and to prevent neurodegeneration having a direct impact on apoptosis and the caspase activation cascade [84][85]. Perhaps more importantly, lifestyle interventions can promote cardiovascular health, boosting mitophagy [86][87]. The future could also bring new findings on novel, non-canonical mechanisms of mitochondrial quality control, such as the recently described mitochondrion-derived vesicles, characterized in hypoxic neurons and cardiomyocytes [88].
References
- Inmaculada Martínez-Reyes; Navdeep S. Chandel; Mitochondrial TCA cycle metabolites control physiology and disease. Nature Communications 2020, 11, 1-11, 10.1038/s41467-019-13668-3.
- John J. Lemasters; Selective Mitochondrial Autophagy, or Mitophagy, as a Targeted Defense Against Oxidative Stress, Mitochondrial Dysfunction, and Aging. Rejuvenation Research 2005, 8, 3-5, 10.1089/rej.2005.8.3.
- Insil Kim; Sara Rodriguez-Enriquez; John J. Lemasters; Selective degradation of mitochondria by mitophagy. Archives of Biochemistry and Biophysics 2007, 462, 245-253, 10.1016/j.abb.2007.03.034.
- Daniela Bakula; Morten Scheibye-Knudsen; MitophAging: Mitophagy in Aging and Disease. Frontiers in Cell and Developmental Biology 2020, 8, 239, 10.3389/fcell.2020.00239.
- Sarah Pickles; Pierre Vigié; Richard J. Youle; Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Current Biology 2018, 28, R170-R185, 10.1016/j.cub.2018.01.004.
- Damian Gatica; Vikramjit Lahiri; Daniel J. Klionsky; Cargo recognition and degradation by selective autophagy. Nature 2018, 20, 233-242, 10.1038/s41556-018-0037-z.
- Seonghee Jung; Hyeonjeong Jeong; Seong-Woon Yu; Autophagy as a decisive process for cell death. Experimental & Molecular Medicine 2020, 52, 921-930, 10.1038/s12276-020-0455-4.
- Tomotake Kanki; Daniel J. Klionsky; Mitophagy in Yeast Occurs through a Selective Mechanism. Journal of Biological Chemistry 2008, 283, 32386-32393, 10.1074/jbc.m802403200.
- Kei Okatsu; Keiko Saisho; Midori Shimanuki; Kazuto Nakada; Hiroshi Shitara; Yu-Shin Sou; Mayumi Kimura; Shigeto Sato; Nobutaka Hattori; Masaaki Komatsu; et al.Keiji TanakaNoriyuki Matsuda p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes to Cells 2010, 15, 887-900, 10.1111/j.1365-2443.2010.01426.x.
- Konstantinos Palikaras; Eirini Lionaki; Nektarios Tavernarakis; Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nature 2018, 20, 1013-1022, 10.1038/s41556-018-0176-2.
- Maria Zachari; Nicholas T. Ktistakis; Mammalian Mitophagosome Formation: A Focus on the Early Signals and Steps. Frontiers in Cell and Developmental Biology 2020, 8, 171, 10.3389/fcell.2020.00171.
- Thomas G. McWilliams; Alan R. Prescott; Lambert Montava-Garriga; Graeme Ball; François Singh; Erica Barini; Miratul M.K. Muqit; Simon P. Brooks; Ian G. Ganley; Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metabolism 2018, 27, 439-449.e5, 10.1016/j.cmet.2017.12.008.
- John J. Lemasters; Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biology 2014, 2, 749-754, 10.1016/j.redox.2014.06.004.
- Jee-Hyun Um; Young Yeon Kim; Toren Finkel; Jeanho Yun; Sensitive Measurement of Mitophagy by Flow Cytometry Using the pH-dependent Fluorescent Reporter mt-Keima. Journal of Visualized Experiments 2018, -, 58099, 10.3791/58099.
- Nuo Sun; Jeanho Yun; Jie Liu; Daniela Malide; Chengyu Liu; Ilsa I. Rovira; Kira M. Holmström; Maria M. Fergusson; Young Hyun Yoo; Christian A. Combs; et al.Toren Finkel Measuring In Vivo Mitophagy. Molecular Cell 2015, 60, 685-696, 10.1016/j.molcel.2015.10.009.
- Nadia Cummins; Jürgen Götz; Shedding light on mitophagy in neurons: what is the evidence for PINK1/Parkin mitophagy in vivo?. Cellular and Molecular Life Sciences 2017, 75, 1151-1162, 10.1007/s00018-017-2692-9.
- Yurong Zhang; Mengdi Zhang; Wei Zhu; Jie Yu; Qiaoyun Wang; Jinjin Zhang; Yaru Cui; Xiaohong Pan; Xue Gao; Hongliu Sun; et al. Succinate accumulation induces mitochondrial reactive oxygen species generation and promotes status epilepticus in the kainic acid rat model. Redox Biology 2020, 28, 101365, 10.1016/j.redox.2019.101365.
- J P Bernardini; M Lazarou; G Dewson; Parkin and mitophagy in cancer. Oncogene 2016, 36, 1315-1327, 10.1038/onc.2016.302.
- Jose Manuel Bravo-San Pedro; Guido Kroemer; Lorenzo Galluzzi; Autophagy and Mitophagy in Cardiovascular Disease. Circulation Research 2017, 120, 1812-1824, 10.1161/circresaha.117.311082.
- Konstantinos Palikaras; Nektarios Tavernarakis; Regulation and roles of mitophagy at synapses. Mechanisms of Ageing and Development 2020, 187, 111216, 10.1016/j.mad.2020.111216.
- Alfonso Schiavi; Flavie Strappazzon; Natascia Ventura; Mitophagy and iron: two actors sharing the stage in age-associated neuronal pathologies. Mechanisms of Ageing and Development 2020, 188, 111252, 10.1016/j.mad.2020.111252.
- Akinori Eiyama; Koji Okamoto; PINK1/Parkin-mediated mitophagy in mammalian cells. Current Opinion in Cell Biology 2015, 33, 95-101, 10.1016/j.ceb.2015.01.002.
- Shiori Sekine; PINK1 import regulation at a crossroad of mitochondrial fate.. The Journal of Biochemistry 2019, 167, 217-224, 10.1093/jb/mvz069.
- Derek Narendra; Atsushi Tanaka; Der-Fen Suen; Richard J. Youle; Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. Journal of Cell Biology 2008, 183, 795-803, 10.1083/jcb.200809125.
- Noriyuki Matsuda; Phospho-ubiquitin: upending the PINK–Parkin–ubiquitin cascade. The Journal of Biochemistry 2016, 159, 379-385, 10.1093/jb/mvv125.
- Nickie C. Chan; Anna M. Salazar; Anh H. Pham; Michael J. Sweredoski; Natalie J. Kolawa; Robert L.J. Graham; Sonja Hess; David C. Chan; Broad activation of the ubiquitin–proteasome system by Parkin is critical for mitophagy. Human Molecular Genetics 2011, 20, 1726-1737, 10.1093/hmg/ddr048.
- Baris Bingol; Morgan Sheng; Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radical Biology and Medicine 2016, 100, 210-222, 10.1016/j.freeradbiomed.2016.04.015.
- Hong-Min Ni; Jessica A. Williams; Wen-Xing Ding; Mitochondrial dynamics and mitochondrial quality control. Redox Biology 2015, 4, 6-13, 10.1016/j.redox.2014.11.006.
- Michael Lazarou; Seok Min Jin; Lesley A. Kane; Richard J. Youle; Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Developmental Cell 2012, 22, 320-333, 10.1016/j.devcel.2011.12.014.
- Emma Deas; Helene Plun-Favreau; Sonia Gandhi; Howard Desmond; Svend Kjaer; Samantha H.Y. Loh; Alan E. Renton; Robert J. Harvey; Alexander J. Whitworth; L. Miguel Martins; et al.Andrey AbramovNicholas W. Wood PINK1 cleavage at position A103 by the mitochondrial protease PARL. Human Molecular Genetics 2010, 20, 867-879, 10.1093/hmg/ddq526.
- Koji Yamano; Richard J Youle; PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758-1769, 10.4161/auto.24633.
- Alicia Pickrell; Richard J. Youle; The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257-273, 10.1016/j.neuron.2014.12.007.
- Verónica Eisner; Martin Picard; György Hajnóczky; Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nature 2018, 20, 755-765, 10.1038/s41556-018-0133-0.
- Angelika S. Rambold; Brenda Kostelecky; Natalie Elia; Jennifer Lippincott-Schwartz; Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proceedings of the National Academy of Sciences 2011, 108, 10190-10195, 10.1073/pnas.1107402108.
- Ligia C. Gomes; Giulietta Di Benedetto; Luca Scorrano; During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nature 2011, 13, 589-598, 10.1038/ncb2220.
- Laura M. Westrate; Jeffrey A. Drocco; Katie R. Martin; William S. Hlavacek; Jeffrey P. MacKeigan; Mitochondrial Morphological Features Are Associated with Fission and Fusion Events. PLOS ONE 2014, 9, e95265, 10.1371/journal.pone.0095265.
- YanShuang Zhou; Qi Long; Xingguo Liu; A new sight: topology-dependent mitophagy. Cell Biology and Toxicology 2020, 36, 199-204, 10.1007/s10565-020-09534-4.
- Pablo E. Morales; Carla Arias-Durán; Yáreni Ávalos-Guajardo; Geraldine Aedo; Hugo E. Verdejo; Valentina Parra; Sergio Lavandero; Emerging role of mitophagy in cardiovascular physiology and pathology. Molecular Aspects of Medicine 2020, 71, 100822, 10.1016/j.mam.2019.09.006.
- Prashant Mishra; David C. Chan; Metabolic regulation of mitochondrial dynamics. Journal of Cell Biology 2016, 212, 379-387, 10.1083/jcb.201511036.
- Jan Dudek; Role of Cardiolipin in Mitochondrial Signaling Pathways. Frontiers in Cell and Developmental Biology 2017, 5, 90-90, 10.3389/fcell.2017.00090.
- Seungyoon B. Yu; Gulcin Pekkurnaz; Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. Journal of Molecular Biology 2018, 430, 3922-3941, 10.1016/j.jmb.2018.07.027.
- Oliver C. Losón; Zhiyin Song; Hsiuchen Chen; David C. Chan; Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Molecular Biology of the Cell 2013, 24, 659-667, 10.1091/mbc.e12-10-0721.
- Jonathan R. Friedman; Laura L. Lackner; Matthew West; Jared R. DiBenedetto; Jodi Nunnari; Gia K. Voeltz; ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358-362, 10.1126/science.1207385.
- Mafalda Escobar-Henriques; Mariana Joaquim; Mitofusins: Disease Gatekeepers and Hubs in Mitochondrial Quality Control by E3 Ligases. Frontiers in Physiology 2019, 10, 517, 10.3389/fphys.2019.00517.
- Atsushi Tanaka; Megan M. Cleland; Shan Xu; Derek P. Narendra; Der-Fen Suen; Mariusz Karbowski; Richard J. Youle; Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. Journal of Cell Biology 2010, 191, 1367-1380, 10.1083/jcb.201007013.
- Yun Chen; Gerald W. Dorn Ii; PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471-475, 10.1126/science.1231031.
- Amadou K. S. Camara; Yifan Zhou; Po-Chao Wen; Emad Tajkhorshid; Wai-Meng Kwok; Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Frontiers in Physiology 2017, 8, 460, 10.3389/fphys.2017.00460.
- Sven Geisler; Kira M. Holmström; Diana Skujat; Fabienne C. Fiesel; Oliver C. Rothfuss; Philipp J. Kahle; Wolfdieter Springer; PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature 2010, 12, 119-131, 10.1038/ncb2012.
- Su Jin Ham; Daewon Lee; Heesuk Yoo; Kyoungho Jun; Heejin Shin; Jongkyeong Chung; Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proceedings of the National Academy of Sciences 2020, 117, 4281-4291, 10.1073/pnas.1909814117.
- Jemma Gatliff; Daniel East; James Crosby; Rosella Abeti; Robert Harvey; William Craigen; Peter Parker; Michelangelo Campanella; TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10, 2279-2296, 10.4161/15548627.2014.991665.
- Xinnan Wang; Dominic Winter; Ghazaleh Ashrafi; Julia Schlehe; Yao Liang Wong; Dennis Selkoe; Sarah Rice; Judith Steen; Matthew J. LaVoie; Thomas L. Schwarz; et al. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell 2011, 147, 893-906, 10.1016/j.cell.2011.10.018.
- Lucia Barazzuol; Flavia Giamogante; Marisa Brini; Tito Calì; PINK1/Parkin Mediated Mitophagy, Ca2+ Signalling, and ER–Mitochondria Contacts in Parkinson’s Disease. International Journal of Molecular Sciences 2020, 21, 1772, 10.3390/ijms21051772.
- Xinnan Wang; Thomas L. Schwarz; The Mechanism of Ca2+-Dependent Regulation of Kinesin-Mediated Mitochondrial Motility. Cell 2009, 136, 163-174, 10.1016/j.cell.2008.11.046.
- Dzhamilja Safiulina; Malle Kuum; Vinay Choubey; Nana Gogichaishvili; Joanna Liiv; Miriam A Hickey; Michal Cagalinec; Merle Mandel; Akbar Zeb; Mailis Liiv; et al.Allen Kaasik Miro proteins prime mitochondria for Parkin translocation and mitophagy. The EMBO Journal 2018, 38, e99384, 10.15252/embj.201899384.
- Paul A. Ney; Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2015, 1853, 2775-2783, 10.1016/j.bbamcr.2015.02.022.
- Terje Johansen; Trond Lamark; Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. Journal of Molecular Biology 2020, 432, 80-103, 10.1016/j.jmb.2019.07.016.
- Michael Lazarou; Danielle A. Sliter; Lesley A. Kane; Shireen Sarraf; Chunxin Wang; Jonathon L. Burman; Dionisia P. Sideris; Adam I. Fogel; Richard J. Youle; The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309-314, 10.1038/nature14893.
- Saori R. Yoshii; Noboru Mizushima; Autophagy machinery in the context of mammalian mitophagy. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2015, 1853, 2797-2801, 10.1016/j.bbamcr.2015.01.013.
- Benjamin Richter; Danielle A. Sliter; Lina Herhaus; Alexandra Stolz; Chunxin Wang; Petra Beli; Gabriele Zaffagnini; Philipp Wild; Sascha Martens; Sebastian A. Wagner; et al.Richard J. YouleIvan Dikic Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proceedings of the National Academy of Sciences 2016, 113, 4039-4044, 10.1073/pnas.1523926113.
- Andrew S. Moore; Erika L. F. Holzbaur; Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proceedings of the National Academy of Sciences 2016, 113, E3349-E3358, 10.1073/pnas.1523810113.
- Rubén Gómez-Sánchez; Sokhna M S Yakhine-Diop; José M Bravo-San Pedro; Elisa Pizarro-Estrella; Mario Rodríguez-Arribas; Vicente Climent; Francisco E Martin-Cano; María E González-Soltero; Anurag Tandon; José M Fuentes; et al.Rosa A González-Polo PINK1 deficiency enhances autophagy and mitophagy induction. Molecular & Cellular Oncology 2015, 3, e1046579, 10.1080/23723556.2015.1046579.
- J Zhang; P A Ney; Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death & Differentiation 2009, 16, 939-946, 10.1038/cdd.2009.16.
- Chengyuan Tang; Hailong Han; Zhiwen Liu; Yuxue Liu; Lijun Yin; Juan Cai; Liyu He; Yu Liu; Guochun Chen; Zhuohua Zhang; et al.Xiao-Ming YinZheng Dong Activation of BNIP3-mediated mitophagy protects against renal ischemia–reperfusion injury. Cell Death & Disease 2019, 10, 1-15, 10.1038/s41419-019-1899-0.
- Lei Liu; Du Feng; Guo Chen; Ming Chen; Qiaoxia Zheng; Pingping Song; Qi Ma; Chongzhuo Zhu; Rui Wang; Wanjun Qi; et al.Lei HuangPeng XueBaowei LiXiaohui WangHaijing JinJun WangFuquan YangPingsheng LiuYushan ZhuSenfang SuiQuan Chen Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature 2012, 14, 177-185, 10.1038/ncb2422.
- Sungwoo Park; Seon-Guk Choi; Seung-Min Yoo; Jin H Son; Yong-Keun Jung; Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy 2014, 10, 1906-1920, 10.4161/auto.32177.
- Koji Yamano; Adam I Fogel; Chunxin Wang; Alexander M Van Der Bliek; Richard J Youle; Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 2014, 3, e01612, 10.7554/elife.01612.
- Xue Xia; Sarah Katzenell; Erin F. Reinhart; Katherine M. Bauer; Maria Pellegrini; Michael J. Ragusa; A pseudo-receiver domain in Atg32 is required for mitophagy. Autophagy 2018, 14, 1620-1628, 10.1080/15548627.2018.1472838.
- Zambarlal Bhujabal; Åsa B Birgisdottir; Eva Sjøttem; Hanne B Brenne; Aud Øvervatn; Sabrina Habisov; Vladimir Kirkin; Trond Lamark; Terje Johansen; FKBP8 recruits LC3A to mediate Parkin‐independent mitophagy. EMBO reports 2017, 18, 947-961, 10.15252/embr.201643147.
- Yongjie Wei; Wei-Chung Chiang; Rhea Sumpter; Prashant Mishra; Beth Levine; Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224-238.e10, 10.1016/j.cell.2016.11.042.
- Charleen Chu; Jing Ji; Ruben K. Dagda; Jian Fei Jiang; Yulia Tyurina; Oleksandr Kapralov; Vladimir A. Tyurin; Naveena Yanamala; Indira H. Shrivastava; Dariush Mohammadyani; et al.Kent Zhi Qiang WangJianhui ZhuJudith Klein-SeetharamanKrishnakumar BalasubramanianAndrew AmoscatoGrigory BorisenkoZhentai HuangAaron M. GusdonAmin CheikhiErin K. SteerRuth WangCatherine BatySimon WatkinsIvet BaharHülya BayırValerian E. Kagan Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature 2013, 15, 1197-1205, 10.1038/ncb2837.
- Alex P. Seabright; Nicholas H. F. Fine; Jonathan P. Barlow; Samuel O. Lord; Ibrahim Musa; Alexander Gray; Jack A. Bryant; Manuel Banzhaf; Gareth G. Lavery; D. Grahame Hardie; et al.David J. HodsonAndrew PhilpYu‐Chiang Lai AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1‐Parkin independent manner. The FASEB Journal 2020, 34, 6284-6301, 10.1096/fj.201903051r.
- Toshiro Saito; Jihoon Nah; Shin-Ichi Oka; Risa Mukai; Yoshiya Monden; Yasuhiro Maejima; Yoshiyuki Ikeda; Sebastiano Sciarretta; Tong Liu; Hong Li; et al.Erdene BaljinnyamDiego FraidenraichLuke FritzkyPeiyong ZhaiShizuko IchinoseMitsuaki IsobeChiao-Po HsuMondira KunduJunichi Sadoshima An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. Journal of Clinical Investigation 2019, 129, 802-819, 10.1172/jci122035.
- Ersheng Kuang; Jianfei Qi; Ze’Ev Ronai; Emerging roles of E3 ubiquitin ligases in autophagy. Trends in Biochemical Sciences 2013, 38, 453-460, 10.1016/j.tibs.2013.06.008.
- Vinay Choubey; Michal Cagalinec; Joanna Liiv; Dzhamilja Safiulina; Miriam Hickey; Malle Kuum; Mailis Liiv; Tahira Anwar; Eeva-Liisa Eskelinen; Allen Kaasik; et al. BECN1 is involved in the initiation of mitophagy. Autophagy 2014, 10, 1105-1119, 10.4161/auto.28615.
- Maria Zachari; Sigurdur R. Gudmundsson; Ziyue Li; Maria Manifava; Fiorella Cugliandolo; Ronak Shah; Matthew Smith; James Stronge; Eleftherios Karanasios; Caterina Piunti; et al.Chieko Kishi-ItakuraHelena VihinenEija JokitaloJun-Lin GuanFolma BussAndrew M. SmithSimon A. WalkerEeva-Liisa EskelinenNicholas T. Ktistakis Selective Autophagy of Mitochondria on a Ubiquitin-Endoplasmic-Reticulum Platform. Developmental Cell 2019, 50, 627-643.e5, 10.1016/j.devcel.2019.06.016.
- Sonia Gandhi; Alison Wood-Kaczmar; Zhi Yao; Helene Plun-Favreau; Emma Deas; Kristina Klupsch; Julian Downward; David S. Latchman; Sarah Tabrizi; Nicholas W. Wood; et al.Michael DuchenAndrey Y. Abramov PINK1-Associated Parkinson's Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Molecular Cell 2009, 33, 627-638, 10.1016/j.molcel.2009.02.013.
- Bavo Heeman; Chris Van Den Haute; Sarah-Ann Aelvoet; Federica Valsecchi; Richard J. Rodenburg; Veerle Reumers; Zeger Debyser; Geert Callewaert; Werner J. H. Koopman; Peter H. G. M. Willems; et al.Veerle Baekelandt Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. Journal of Cell Science 2011, 124, 1115-1125, 10.1242/jcs.078303.
- Rubén Gómez-Sánchez; Matthew E. Gegg; Jose Manuel Bravo-San Pedro; Mireia Niso-Santano; Lydia Alvarez-Erviti; Elisa Pizarro-Estrella; Yolanda Gutiérrez-Martín; Alberto Alvarez-Barrientos; José M. Fuentes; Rosa Ana González-Polo; et al.Anthony H.V. Schapira Mitochondrial impairment increases FL-PINK1 levels by calcium-dependent gene expression. Neurobiology of Disease 2014, 62, 426-440, 10.1016/j.nbd.2013.10.021.
- Vania Gelmetti; Priscilla De Rosa; Liliana Torosantucci; Elettra Sara Marini; Alessandra Romagnoli; Martina Di Rienzo; Giuseppe Arena; Domenico Vignone; Gian Maria Fimia; Enza Maria Valente; et al. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654-669, 10.1080/15548627.2016.1277309.
- Ming Yang; Chenrui Li; Shikun Yang; Ying Xiao; Xiaofen Xiong; Wei Chen; Hao Zhao; Qin Zhang; Yachun Han; Lin Sun; et al. Mitochondria-Associated ER Membranes – The Origin Site of Autophagy. Frontiers in Cell and Developmental Biology 2020, 8, 595, 10.3389/fcell.2020.00595.
- Junyang Jung; Youngbuhm Huh; Ulfuara Shefa; Na Young Jeong; In Ok Song; Hyung-Joo Chung; Dokyoung Kim; Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regeneration Research 2019, 14, 749-756, 10.4103/1673-5374.249218.
- Alessandra Stacchiotti; Giovanni Corsetti; Natural Compounds and Autophagy: Allies Against Neurodegeneration. Frontiers in Cell and Developmental Biology 2020, 8, 555409, 10.3389/fcell.2020.555409.
- Nimmy Varghese; Selina Werner; Amandine Grimm; Anne Eckert; Dietary Mitophagy Enhancer: A Strategy for Healthy Brain Aging?. Antioxidants 2020, 9, 932, 10.3390/antiox9100932.
- Yan Zhao; Shaohui Huang; Jie Liu; Ximing Wu; Shuai Zhou; Ke Dai; Yurong Kou; Mitophagy Contributes to the Pathogenesis of Inflammatory Diseases. Inflammation 2018, 41, 1590-1600, 10.1007/s10753-018-0835-2.
- Jae-Min Yuk; Prashanta Silwal; Eun-Kyeong Jo; Inflammasome and Mitophagy Connection in Health and Disease. International Journal of Molecular Sciences 2020, 21, 4714, 10.3390/ijms21134714.
- Giampaolo Morciano; Simone Patergnani; Massimo Bonora; Gaia Pedriali; Anna Tarocco; Esmaa Bouhamida; Saverio Marchi; Gina Ancora; Gabriele Anania; Mariusz R. Wieckowski; et al.Carlotta GiorgiPaolo Pinton Mitophagy in Cardiovascular Diseases. Journal of Clinical Medicine 2020, 9, 892, 10.3390/jcm9030892.
- Ne N. Wu; Haili Tian; Peijie Chen; Dan Wang; Jun Ren; Yingmei Zhang; Physical Exercise and Selective Autophagy: Benefit and Risk on Cardiovascular Health. Cells 2019, 8, 1436, 10.3390/cells8111436.
- Binghu Li; Hongliang Zhao; Yue Wu; Yu Zhu; Jie Zhang; Guangming Yang; Qingguang Yan; Junxia Li; Tao Li; Liangming Liu; et al. Mitochondrial-Derived Vesicles Protect Cardiomyocytes Against Hypoxic Damage. Frontiers in Cell and Developmental Biology 2020, 8, 214, 10.3389/fcell.2020.00214.
- Ne N. Wu; Haili Tian; Peijie Chen; Dan Wang; Jun Ren; Yingmei Zhang; Physical Exercise and Selective Autophagy: Benefit and Risk on Cardiovascular Health. Cells 2019, 8, 1436, 10.3390/cells8111436.
- Binghu Li; Hongliang Zhao; Yue Wu; Yu Zhu; Jie Zhang; Guangming Yang; Qingguang Yan; Junxia Li; Tao Li; Liangming Liu; et al. Mitochondrial-Derived Vesicles Protect Cardiomyocytes Against Hypoxic Damage. Frontiers in Cell and Developmental Biology 2020, 8, 214, 10.3389/fcell.2020.00214.