 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fanni Tóth | + 8716 word(s) | 8716 | 2021-01-07 05:23:54 | | | |
| 2 | Dean Liu | -5334 word(s) | 3382 | 2021-05-27 08:54:55 | | |
Video Upload Options
Some natural products and molecules are very promising neuroprotective agents because of their structural diversity and wide variety of biological activities. In addition to their neuroprotective effect, they are known for their antioxidant, anti-inflammatory and antiapoptotic effects and often serve as a starting point for drug discovery.
1. Introduction
Neuronal damage in the central nervous system (CNS) is universal in neurodegenerative diseases (NDs) [1]. NDs are defined by the progressive loss of neurons in the CNS, which generates deficits in brain function [2]. The symptoms of NDs vary from memory and cognitive deficits to the deterioration of one’s capability to breath or move [3]. The most frequent NDs are Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS). The incidence of NDs has increased greatly worldwide due to the rise in life expectancy, and this associates them with profound social and economic burdens [4].
The molecular mechanisms of neuronal damage are mostly based on excitatory amino acid release and oxidative stress, causing mitochondrial dysfunction [5] (Figure 1). Under physiological conditions, in the CNS, excitatory amino acids are crucial neurotransmitters and their release and uptake are very well controlled. Nonetheless, their accumulation can lead to brain damage [6]. In glutamate-induced excitotoxicity, glutamate activates N-methyl-d-aspartic acid receptors (NMDARs), leading to a Ca2+ overload [7]. This process is associated with increased reactive oxygen species (ROS), as well as mitochondrial dysfunction resulting in neuronal apoptosis [8][9]. The brain is more vulnerable to damage by oxidative stress due to its high content of polyunsaturated fatty acids [10], that are very prone to ROS attacks [11], which result in lipid peroxidation. Furthermore, the brain has a high rate of O2 utilization and a low antioxidant defense, and the accumulated metals like copper or iron are capable of catalyzing the formation of hydroxyl radicals [12].
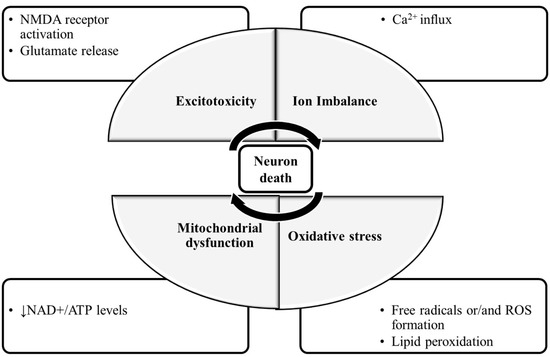
Figure 1. The molecular mechanisms of neuronal damage. The mechanisms of neuronal death are focused on excitatory amino acid release, calcium influx and calcium overload, oxidative stress and mitochondrial dysfunction. During glutamate-induced excitotoxicity, glutamate activates NMDA receptors, leading to a Ca2+ influx and overload, which is associated with increased ROS formation and the damaged mitochondria resulting in neuronal death. Abbreviations: NMDA receptor: N-methyl-d-aspartic acid receptor; NAD+: nicotinamide adenine dinucleotide; ATP: adenosine triphosphate; ROS: reactive oxygen species.
Neuroprotection denotes approaches that defend the CNS against neuronal injury and/or death while subjected to trauma or neurodegenerative disorders. It slows the progression of the disease and prevents neuronal death [13]. Hence, neuroprotection is a crucial part of care for NDs [14].
Neuroprotection can be classified into three groups: pharmacological-, non-pharmacological- and cellular and genetic approaches. Pharmacological approaches include antioxidants, neurotransmitter agonists/antagonists, anti-inflammatory drugs and natural products [15]. Non-pharmacological approaches include exercise that influences body metabolism [16], diet control to reduce risk factors such as hyperlipidemia [17] and acupuncture, that can help adjust body metabolism and immunity [18]. Cellular and genetic approaches include growth/trophic factors [19].
Considering there are various changes that occur in the aging brain, it is implausible that targeting a single change is able to intervene in the complexity of the disease progression. Hence, compounds with multiple biological activities affecting the different age-associated factors that contribute to ND development and progression are extremely needed [20]. The existing therapies available for NDs only relieve symptoms [21].
Natural products (including natural molecules) are defined as organic compounds synthesized by living organisms. Some of them are very promising neuroprotective agents because of their structural diversity and wide variety of biological activities [4]. The major neuroprotective targets of natural products and molecules are excitotoxicity, apoptosis, mitochondrial dysfunction, inflammation, oxidative stress and protein misfolding [22][23]. They have anti-neurodegenerative, antioxidant, anti-inflammatory and antiapoptotic effects [24][25]. Natural products and molecules are commonly used as starting points for drug discovery, from which synthetic analogs are synthetized to improve efficacy, potency and to reduce side effects and to increase bioavailability. A lot of the U.S. Food and Drug Administration (FDA)-approved drugs are prompted by natural products [26].
In this review, the following natural molecules are discussed: kynurenic acid, pantethine and α-lipoic acid. Common features of all three natural molecules that they are (i) neuroprotective (ii) antioxidant (iii) reducing agents, which have important roles in glycolysis and the TCA cycle (Figure 2). This makes them potential candidates for neuroprotective therapies for managing NDs.
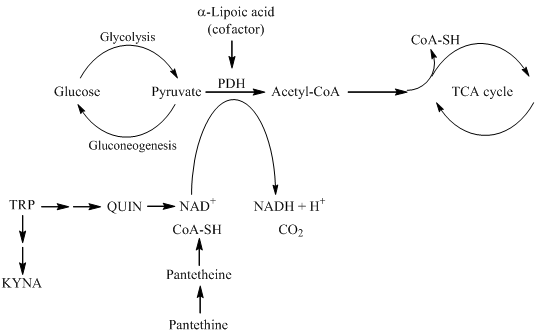
Figure 2. The roles of kynurenine pathway metabolites, pantethine and α-lipoic acid in glycolysis and TCA cycle. PDH is the link between glycolysis and the TCA cycle. α-lipoic acid functions as a cofactor for pyruvate dehydrogenase. The kynurenine pathway of TRP metabolism ultimately leads to the formation of NAD+, which will be reduced to NADH. Pantethine is a precursor in the formation of CoA. which functions as an acetyl carrier. It transfers acetyl groups from pyruvate to oxaloacetate, initiating the TCA cycle. TRP tryptophan; KYNA kynurenic acid; QUIN quinolinic acid; NAD+ nicotinamide adenine dinucleotide; CoA coenzyme A; TCA tricarboxylic acid; PDH pyruvate dehydrogenase.
2. Kynurenic Acid
Kynurenines are considered a hot topic nowadays, as in the last 20 years (2000–2020) more than 4600 articles have been published on the topic [27].
Tryptorphan (TRP) is an essential amino acid, a building block for protein synthesis and also a precursor for the synthesis of serotonin, kynurenic acid (KYNA) and nicotinamide adenine dinucleotide (NAD+). The main metabolic route of TRP degradation is through the kynurenine pathway (KP). More than 95% of TRP is metabolized through this route, and only 5% is degraded through the methoxy-indole pathway [28]. The KP is activated by free radicals, interferons and cytokines, which induce the activity of indoleamine 2,3-dioxygenase (IDO) [29] and tryptophan 2,3-dioxygenase (TDO) enzymes [30], the rate-limiting enzymes of the pathway [31].
The KP is comprised of several enzymatic steps (Figure 3), which ultimately lead to the formation of NAD+, which has a pivotal role in different cellular functions (energy metabolism, gene expression, cell death and regulation of calcium homeostasis) [32]. KP’s center compound is L-kynurenine (KYN) [33], which can be further degraded through three different routes, resulting in several neuroactive metabolites. KYNA, the main neuroprotective agent is formed after KYN is catalyzed by the enzyme kynurenine aminotransferase (KAT) [34], whereas quinolinic acid (QUIN) [35] and 3-hydroxy-L-kynurenine (3HK) [36] both show neurotoxic properties.
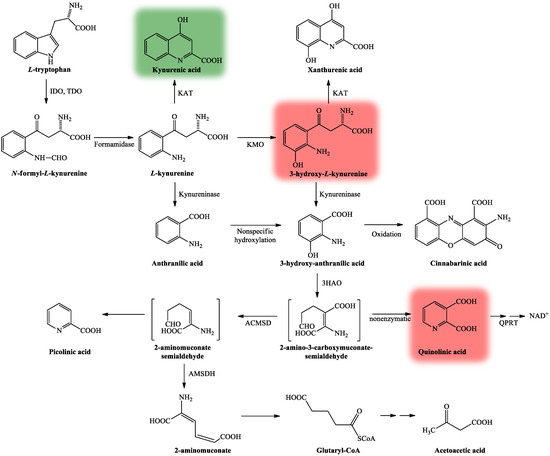
Figure 3. The kynurenine pathway. More than 95% of TRP is metabolized through the KP [28]. The L-tryptophan converting IDO and TDO depict the rate-limiting enzymes of the pathway [31]. KP’s center metabolite is L-kynurenine [33] which can be further degraded through three distinct routes to form several neuroactive metabolites: kynurenic acid, 3-hydroxy-L-kynurenine and quinolinic acid, which are cardinal in the CNS. Kynurenic acid is formed from L-kynurenine in astrocytes and neurons by KATs [34]. Quinolinic acid and 3-hydroxy-L-kynurenine are synthesized by infiltrating macrophages and microglia [35]. Neurotoxicity is mediated by quinolinic acid and 3-hydroxy-L-kynurenine (color red) via NMDA receptor agonism and free radical production [36], while neuroprotection can be exerted by kynurenic acid (color green) by acting as an antagonist at the NMDA receptor [37]. Abbreviations: TRP: tryptophan; KP: kynurenine pathway; CNS: central nervous system; NMDA receptor: N-methyl-d-aspartic acid receptor; 3HAO: 3-hydroxyanthranilate oxidase; ACMSD: 2-amino-3-carboxymuconate-semialdehyde decarboxylase; AMSDH: 2-aminomuconate-6-semialdehyde dehydrogenase; IDO/TDO: indoleamine 2,3-dioxygenase/tryptophan 2,3-dioxygenase; KAT: kynurenine aminotransferase; KMO: kynurenine 3-monooxygenase; NAD+: nicotinamide adenine dinucleotide; QPRT: quinolinic acid phosphoribosyltransferase.
In the CNS, KYNA acts at multiple receptors. KYNA is an endogenous competitive antagonist with high affinity at the strychnine-insensitive glycine-binding NR1 site of NMDARs, it exerts antidepressant and psychotomimetic effects [37,38]. KYNA can bind to the NMDA recognition NR2 site of the receptor as well, albeit with a weaker affinity [39,40]. It also acts upon α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors via two distinct mechanisms: at low (nanomolar to micromolar) concentrations it facilitates AMPA receptor responses, whereas at high (millimolar) concentrations it competitively antagonizes glutamate receptors [41,42]. KYNA exerts an endogenous agonistic effect on the orphan G protein-coupled receptor (GPR35) [43]. Additionally, KYNA is an endogenous agonist at the aryl hydrocarbon receptor (AHR), expressed in immune cells and in tumor cells [44,45] (Table 1). In 2001, it was suggested by Hilmas et al. that KYNA is a noncompetitive inhibitor of the α7 nicotinic acetylcholine receptor (α-7nAChR) [46], however this hypothesis is much debated. The current standpoint is that it does not directly affect nicotinic receptors and results with it should only be explained by KYNA’s confirmed sites of action [47].
Table 1. Main binding sites of kynurenic acid
|
Receptor |
Action |
|
AHR |
Agonist |
|
GPR35 |
Agonist |
|
NMDAR (glycine-2 co agonist NR1 site) |
Antagonist |
|
NMDAR (glutamate/ NMDA NR2 site) |
Antagonist |
|
AMPAR |
Agonist/Antagonist (dose-dependent) |
GPR35 G protein-coupled receptor 35; AHR aryl hydrocarbon receptor; NMDAR N-methyl-D-aspartic acid receptor; AMPAR α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor.
Since KYNA acts upon multiple receptors, an abnormal decrease or increase in its level may disrupt the equilibrium of neurotransmitter systems, as it can be seen in various neurodegenerative- and neuropsychiatric disorders (Table 2). KYNA could have therapeutic importance for neurological disorders [48], but since its ability to cross the blood-brain barrier (BBB) is limited [49], its use as a neuroprotective agent is somewhat limited. One option to prevent neurodegenerative diseases, which includes the influence of the metabolism towards the neuroprotective branch of the KP. Three potential therapeutic strategies for drug development are known: (i) KYNA analogs with better bioavailability and higher affinity to the binding sites of excitatory receptors; (ii) prodrugs of KYNA, which easily cross the BBB combined with an organic acid transport inhibitor to increase brain KYNA levels and (iii) inhibitors of enzymes of the KP [50].
Table 2. Kynurenic acid and kynurenine aminotransferase alterations in neurological diseases
|
Alzheimer’s disease plasma serum, erythrocytes CSF |
KYNA ↓ KYNA ↓ KYNA ↓ |
|
Parkinson’s disease frontal cortex, putamen (L-dopa treatment) plasma serum |
KYNA ↓ KYNA ↓ KAT I, KAT II ↓ |
|
Huntington’s disease cortex striatum CSF |
KYNA ↓ KYNA ↓, KAT ↓ KYNA ↓ |
|
Amyotrophic lateral sclerosis CSF (patients with bulbar onset or severe clinical status) |
KYNA ↑ |
|
Multiple sclerosis plasma erythrocytes CSF (patients with acute relapse) CSF (patients with chronic remission) |
KYNA ↑ KAT I, KAT II ↑ KYNA ↑ KYNA ↓ |
Abbreviations: ↓: decrease in level; ↑: increase in level; KYNA kynurenic acid; CSF cerebrospinal fluid; KAT kynurenine aminotransferase.
3. Pantethine
A common feature of pantethine and tryptophan metabolism is that they have a metabolite somehow connected to the TCA cycle, i.e., in the KP route of TRP metabolism, the neurotoxic compound QUIN will lead to the formation of NAD+, whereas pantethine is responsible for the formation of CoA, important in the delivery of the acetyl-group to the tricarboxylic acid (TCA) cycle. Pantethine’s importance was revealed in 1949, when a new compound, named Lactobacillus bulgaricus factor (LBF) was discovered due to its capability to promote the growth of Lactobacillus bulgaricus. LBF was universally distributed in the natural materials [38]. LBF was shown to be a fragment of coenzyme A (CoA) and in the essential growth factor, mercaptoamine was combined with pantothenic acid (Vitamin B5) as an amide [39]. This substance occurs in two forms: pantetheine and pantethine.
Pantetheine is the cysteamine amide analog of pantothenic acid (vitamin B5) and it is an intermediate in the synthesis of CoA. In pantethine, two molecules of pantetheine are linked by a disulfide bridge. This forms the active part of the CoA molecule (Figure 5).
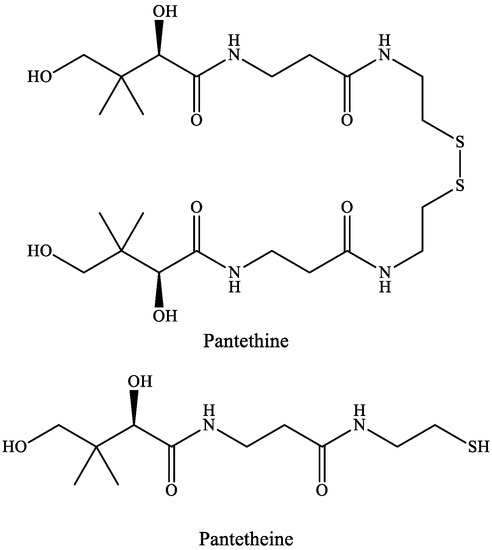
Figure 5. The structure of pantethine and pantetheine.
Most plants and microorganisms can enzymatically combine pantoic acid with β-alanine to produce pantothenic acid. Mammals are not able to synthesize pantothenic acid since they lack the enzyme. Different foods contain CoA, pantethine, pantetheine and pantothenic acid, so the endogenous synthesis of CoA can begin with pantothenic acid.
Regarding the metabolism of pantethine, following oral or intravenous intake, pantethine is immediately hydrolyzed to pantetheine in the small intestine membranes and in blood. Pantetheine can then be phosphorylated to 4′-phosphopantetheine, which is later converted to dephospho-CoA-SH, and finally to CoA-SH in the mitochondria. Pantetheine is transformed to pantothenic acid and cysteamine in hepatocytes. Cysteamine is subsequently metabolized to taurine or is reused to form pantetheine. Pantothenic acid cannot be further degraded in the liver [40][41] (Figure 6).
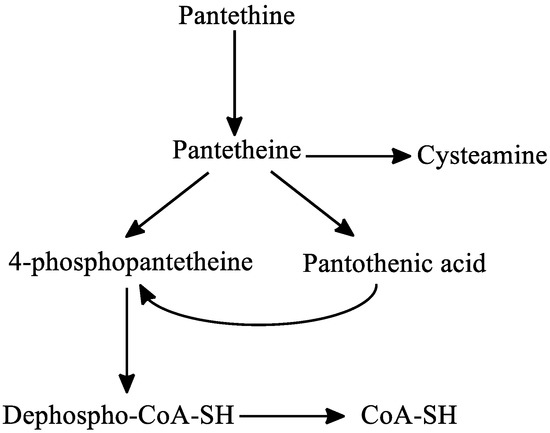
Figure 6. The metabolism of pantethine. Abbreviations: CoA: coenzyme-A.
Since cysteamine in high doses depletes somatostatin and prolactin in different organs, the effect of pantethine was investigated on the level of these hormones in different tissues. Pantethine significantly reduced somatostatin concentrations in the following tissues: duodenal mucosa, gastric mucosa, pancreas, cerebral cortex and hypothalamus [42]. Prolactin was also markedly reduced in the pituitary and in plasma by pantethine, so it can be considered for the management of hyperprolactinemia [43]. In rats, peripherally injected cysteamine and to a lesser extent pantethine reduced noradrenaline and increased dopamine and 3,4-dihydroxyphenylacetic acid (DOPAC) hypothalamic concentrations [44]. Carbon tetrachloride-induced hepatotoxicity in rats is protected by pantethine, pantothenic acid and cystamine. Pantethine provided the greatest protection [45].
3.1. Pantethine and Alzheimer's disease, Parkinson's disease, Major Depressive Disorder and PKAN syndrome
Regarding Alzheimer’s disease (AD) in primary cultured astrocytes of 5XFAD mice, pantethine mitigates metabolic dysfunctions and decreases astrogliosis and IL-1β production [138]. These results associated with pantethine lead to the investigation of its effects in vivo in the 5XFAD (Tg) mouse model of AD. Long-term pantethine treatment significantly reduced glial reactivity and Αβ accumulation, and modulated the aggressive attitude of Tg mice. Based on these results, pantethine could be contemplated as a possible therapeutic option for preventing, slowing, or halting AD progression [139].
In the MPTP-mouse model of Parkinson’s (PD), pantethine reduced MPTP induced neurotoxicity in treated mice, by enhancing fatty acid β-oxidation, which causes an increase in the levels of circulating ketone bodies (KB) and an improvement of mitochondrial function [141]. Pantethine protects from MPTP-induced BBB leakage and significantly mitigates clinical scores [142]. Pantethine has the same effects as KB administration and ketogenic diets, but with multiple advantages, including the prevention of the damaging effect of the long-term administration of high fat diets, due to its hypolipidemic properties [144]. This natural compound should be considered too as a potential therapy against PD.
In the treatment of Major Depressive Disorder (MDD) pantethine may be one of the most promising agents, as it is a naturally occurring substance that can be administered orally with hardly any side effects, and it’s further metabolizes to cysteamine. Another advantage of pantethine is an anti-arteriosclerotic medicine sold by some pharmaceutical companies [152], and many geriatric depression patients may have an arteriosclerotic etiology [153].
Pantothenate kinase-associated neurodegeneration (PKAN) syndrome is the most common form of a group of genetic disorders, called neurodegeneration with brain iron accumulation, which are characterized by iron overload in the brain and are diagnosed by radiological and histopathological examinations [154].
In a PKAN Drosophila model, pantothenate kinase deficiency caused a neurodegenerative phenotype and a reduced lifespan. This Drosophila model revealed that impairment of pantothenate kinase is linked to decreased levels of CoA, mitochondrial dysfunction and increased protein oxidation. Rescue of the phenotype found in the hypomorph mutant dPANK/fbl is obtained by pantethine feeding, which recovers CoA levels, ameliorates mitochondrial function, rescues brain degeneration, improves locomotor abilities, and extends lifespan [159]. The zebrafish orthologue of hPANK2 can be found on chromosome 13. Down-regulation of pank2 can cause a lack of CoA in zebrafish embryos in specific cells and tissues. Compensation of the wild type phenotype can be obtained by exposing P2-MO-injected embryos to 30 μM pantethine [160]. Pank2 knockout mice had structural alteration of muscle morphology, which was similar with that observed in PKAN patients. Pantethine administration was effective in ameliorating the onset of the neuromuscular phenotype observed in Pank2 knockout mice, which were fed a ketogenic diet [155]. Overall, these data indicate that pantethine administration to PKAN patients should be contemplated as a potential, safe and non-toxic therapeutic approach.
4. α-Lipoic Acid
LA has a redox active disulfide group and it is found naturally in mitochondria as the coenzyme for pyruvate dehydrogenase and α-ketoglutarate dehydrogenase. Small amounts of LA are found in foods (spinach and broccoli) and it is also synthesized in the liver [46]. LA was first isolated in 1951 from bovine liver [47]. Dihydrolipoic acid (DHLA), which is the reduced form of LA, interacts with ROS and reactive nitrogen species (RONS) [48]. Both LA and DHLA have antioxidant effects [49]. LA easily penetrates the BBB [50], after which it is quickly internalized by cells and tissues and is reduced to DHLA [51]. LA is active in aqueous or lipophilic [48] environments. Its conjugate base, lipoate, is more soluble and under physiological conditions it is the most common form of LA. It has a highly negative redox potential of −0.32 V [52], therefore, the redox couple LA/DHLA is a “universal antioxidant” (Figure 7.) [53].
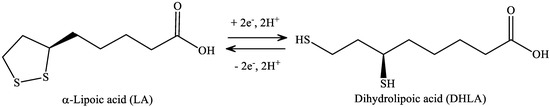
Figure 7. Structure of α-lipoic acid and dihydrolipoic acid.
LA has antioxidant and anti-inflammatory effects [54]. LA supplementation is effective in animal models in obesity and cardiometabolic disorders. LA produces a decrease in body weight [55]. This effect can be clarified by the suppression of protein kinase 5′ adenosine monophosphate-activated protein kinase (AMPK), its hypothalamic action is pivotal for the regulation of food intake and energy expenditure elevation [56]. In animal models, LA supplementation generated the attenuation of ROS and RONS [57], which are both associated with the reduction in lifespan. The oral supplementation of LA can be a potential supplement in cancer treatment, as it improved survival [58] and reduced unwanted effects of chemotherapy [59]. LA is important in combating inflammation and pain. Positive data on rheumatoid arthritis [60], chronic pain [61], neuropathy [62], migraines [63], ulcerative colitis [64] were published.
In various clinical trials which investigated the therapeutic potential of LA, it was concluded that moderate doses (up to 1800 mg/day) were considered safe. Meanwhile, high doses or intraperitoneally administered LA, at a dosage of 5 to 10 g/day, can elevate hydroperoxide levels in the blood [65].
4.1. α-Lipoic acid and Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and Multiple Sclerosis
In an open clinical study, 600 mg LA was given daily to nine patients with AD (receiving a standard treatment with acetylcholinesterase inhibitors) to examine the influence of LA on the progression of AD. LA treatment stabilized cognitive functions in the patients, shown by constant scores in neuropsychological tests (mini-mental state examination, AD assessment scale and cognitive subscale) [196]. LA has the ability to intervene with pathogenic principles of AD, as it stimulates acetylcholine (ACh) production by activating choline acetyltransferase and elevating glucose uptake, as a result providing more acetyl-CoA for ACh production [197]. LA might represent a potential neuroprotective therapy for AD.
In experimental models of PD, lipopolysaccharide (LPS) can be used to activate glial cells [200]. Nasal LPS-induced PD is completely inflammation-driven, and it effectively replicates the chronic, progressive PD pathology [201]. LA can block the LPS-induced inflammatory process [202]. LA administration ameliorated motor dysfunction, preserved dopaminergic neurons and reduced SN α-synuclein accumulation. In M1 microglia LA blocked nuclear factor-κB activation and expression of pro-inflammatory molecules. Further neuroprotective action of LA was studied in an experimental model of PD, induced in male Wistar rats by intrastriatal injection of 6-OHDA. LA improved learning and memory performance and neuromuscular coordination. LA significantly reduced lipid peroxidation levels, and recovered catalase activity and dopamine levels that were damaged by 6-OHDA administration all which lead to a reduction of oxidative stress [203]. It can be concluded that LA displays significant antiparkinsonian effects.
In the 3-NP rat model of HD the neuroprotective effect of LA and acetyl-L-carnitine (ALCAR) on 3-NP-induced alterations in mitochondrial structure, lipid composition, and memory functions was investigated. The combined supplementation of LA + ALCAR improved mitochondrial lipid composition, blocked mitochondrial structural changes, and mitigated cognitive deficits in 3-NP-treated animals. Thus, a combined supplementation of LA + ALCAR can be a possible therapeutic strategy in HD management [206]. It was examined whether LA exerted neuroprotective effects in transgenic mouse models of HD. LA generated significant increases in survival in R6/2 and N171-82Q transgenic mouse models of HD. These results indicate that LA may have valuable effects in HD patients [207].
Studies investigating the effects of LA on demyelination and axonal damage in optic nerve, spinal cord, and brain reported that LA-treated EAE animals had reduced damage in the CNS, which was timing- and route of administration dependent [211,213,214]. LA administration by intraperitoneal (i.p.) injection seven days or directly after immunization protected axons from demyelination and damage [211,213]. Delayed LA administration also decreased damage to the optic nerve but not as profoundly as the immediate treatment [213]. Oral administration was only protective immediately, but not delayed after EAE immunization [211]. LA treatment lead to a reduced disease severity in EAE model animals [215].
The use of LA is desirable in numerous areas of health. LA has various beneficial effects on the aging process and neurological disorders. More evaluation is needed to instruct health professionals better on the safety of prescribing LA as a supplement.
5. Conclusion
In conclusion, the extensive research and development of natural products including KYNA, pantethine and α-lipoic acid, will lead to information which will potentially enable novel drug discovery. The neuroprotective property of these compounds makes them worthy of much more attention, and contemplates them as a possible therapeutic option for several neurological diseases.
References
- Fei, F.; Su, N.; Li, X.; Fei, Z. Neuroprotection mediated by natural products and their chemical derivatives. Neural Regen. Res. 2020, 15, 2008–2015.
- Angeloni, C.; Vauzour, D. Natural products and neuroprotection. Int. J. Mol. Sci. 2019, 20, 5570.
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118.
- González-Cofrade, L.; de las Heras, B.; Ticona, L.A.; Palomino, O.M. Molecular targets involved in the neuroprotection mediated by terpenoids. Planta Med. 2019, 85, 1304–1315.
- Mattiasson, G.; Shamloo, M.; Gido, G.; Mathi, K.; Tomasevic, G.; Yi, S.; Warden, C.H.; Castilho, R.F.; Melcher, T.; Gonzalez-Zulueta, M.; et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat. Med. 2003, 9, 1062–1068.
- Rama, R.; García, J.C. Excitotoxicity and Oxidative Stress in Acute Stroke. In Ischemic Stroke; Schaller, B., Ed.; IntechOpen: London, UK, 2016; pp. 17–42.
- Choi, D.W. Calcium and excitotoxic neuronal injury. Ann. N. Y. Acad. Sci. 1994, 747, 162–171.
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflügers Arch. Eur. J. Physiol. 2010, 460, 525–542.
- Yildiz-Unal, A.; Korulu, S.; Karabay, A. Neuroprotective strategies against calpain-mediated neurodegeneration. Neuropsychiatr. Dis. Treat. 2015, 11, 297–310.
- Rice-Evans, C.; Burdon, R. Free radical-lipid interactions and their pathological consequences. Prog. Lipid Res. 1993, 32, 71–110.
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289.
- Sakr, H.F.; Abbas, A.M.; El Samanoudy, A.Z. Effect of vitamin E on cerebral cortical oxidative stress and brain-derived neurotrophic factor gene expression induced by hypoxia and exercise in rats. J. Physiol. Pharmacol. 2015, 66, 191–202.
- Cai, W.; Zhang, K.; Li, P.; Zhu, L.; Xu, J.; Yang, B.; Hu, X.; Lu, Z.; Chen, J. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: An aging effect. Ageing Res. Rev. 2017, 34, 77–87.
- Brahmachari, G. Discovery and Development of Neuroprotective Agents from Natural Products: An Overview. In Discovery and Development of Neuroprotective Agents from Natural Products, 1st ed.; Brahmachari, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–7.
- Chang, R.C.C.; Ho, Y.S. (Eds.) Introductory Chapter: Concept of Neuroprotection—A New Perspective. In Neuroprotection, 1st ed.; IntechOpen: London, UK, 2019; pp. 1–9.
- Liu, Y.; Yan, T.; Chu, J.M.-T.; Chen, Y.; Dunnett, S.; Ho, Y.-S.; Wong, G.T.-C.; Chang, R.C.-C. The beneficial effects of physical exercise in the brain and related pathophysiological mechanisms in neurodegenerative diseases. Lab. Invest. 2019, 99, 943–957.
- Appleton, J.P.; Scutt, P.; Sprigg, N.; Bath, P.M. Hypercholesterolaemia and vascular dementia. Clin. Sci. 2017, 131, 1561–1578.
- Cao, Y.; Zhang, L.-W.; Wang, J.; Du, S.-Q.; Xiao, L.-Y.; Tu, J.F.; Liu, C.-Z. Mechanisms of acupuncture effect on Alzheimer’s disease in animal based researches. Curr. Top. Med. Chem. 2016, 16, 574–578.
- Semkova, I.; Krieglstein, J. Neuroprotection mediated via neurotrophic factors and induction of neurotrophic factors. Brain Res. Rev. 1999, 30, 176–188.
- Maher, P. The potential of flavonoids for the treatment of neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 3056.
- Di Paolo, M.; Papi, L.; Gori, F.; Turillazzi, E. Natural products in neurodegenerative diseases: A great promise but an ethical challenge. Int. J. Mol. Sci. 2019, 20, 5170.
- Leonoudakis, D.; Rane, A.; Angeli, S.; Lithgow, G.J.; Andersen, J.K.; Chinta, S.J. Anti-Inflammatory and neuroprotective role of natural product securinine in activated glial cells: Implications for Parkinson’s disease. Mediat. Inflamm. 2017, 2017, 8302636.
- Deshpande, P.; Gogia, N.; Singh, A. Exploring the efficacy of natural products in alleviating Alzheimer’s disease. Neural Regen. Res. 2019, 14, 1321–1329.
- Angeloni, C.; Giusti, L.; Hrelia, S. New neuroprotective perspectives in fighting oxidative stress and improving cellular energy metabolism by oleocanthal. Neural Regen. Res. 2019, 14, 1217–1218.
- Flanagan, E.; Müller, M.; Hornberger, M.; Vauzour, D. Impact of flavonoids on cellular and molecular mechanisms underlying age-related cognitive decline and neurodegeneration. Curr. Nutr. Rep. 2018, 7, 49–57.
- Western Oregon University. Available online: (accessed on 31 October 2020).
- Web of Science. Available online: (accessed on 31 October 2020).
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82.
- Hayaishi, O. Properties and function of indoleamine 2,3-dioxygenase. J. Biochem. 1976, 79, 13–21.
- Schröcksnadel, K.; Wirleitner, B.; Winkler, C.; Fuchs, D. Monitoring tryptophan metabolism in chronic immune activation. Clin. Chim. Acta 2006, 364, 82–90.
- Mackay, G.M.; Forrest, C.M.; Stoy, N.; Christofides, J.; Egerton, M.; Stone, T.W.; Darlington, L.G. Tryptophan metabolism and oxidative stress in patients with chronic brain injury. Eur. J. Neurol. 2006, 13, 30–42.
- Ying, W. NAD+ and NADH in cellular functions and cell death. Front. Biosci. 2006, 11, 3129–3148.
- Wirleitner, B.; Neurauter, G.; Schröcksnadel, K.; Frick, B.; Fuchs, D. Interferon-γ- induced conversion of tryptophan: Immunologic and neuropsychiatric aspects. Curr. Med. Chem. 2003, 10, 1581–1591.
- Guidetti, P.; Amori, L.; Sapko, M.T.; Okuno, E.; Schwarcz, R. Mitochondrial aspartate aminotransferase: A third kynurenate-producing enzyme in the mammalian brain. J. Neurochem. 2007, 102, 103–111.
- Bohár, Z.; Toldi, J.; Fülöp, F.; Vécsei, L. Changing the face of kynurenines and neurotoxicity: Therapeutic considerations. Int. J. Mol. Sci. 2015, 16, 9772–9793.
- Németh, H.; Toldi, J.; Vécsei, L. Role of kynurenines in the central and peripherial nervous systems. Curr. Neurovasc. Res. 2005, 2, 249–260.
- Parsons, C.G.; Danysz, W.; Quack, G.; Hartmann, S.; Lorenz, B.; Wollenburg, C.; Baran, L.; Przegalinski, E.; Kostowski, W.; Krzascik, P.; et al. Novel systemically active antagonists of the glycine site of the N-methyl-D-aspartate receptor: Electrophysiological, biochemical and behavioral characterization. J. Pharmacol. Exp. Ther. 1997, 283, 1264–1275.
- Williams, W.L.; Hoff-Jorgensen, E.; Snell, E.E. Determination and properties of an unidentified growth factor required by Lactobacillus bulgaricus. J. Biol. Chem. 1949, 177, 933–940.
- Snell, E.E.; Brown, G.M. Pantethine and related forms of the Lactobacillus bulgaricus factor (LBF). Adv. Enzymol. Relat. Areas Mol. Biol. 1953, 14, 49–71.
- Horváth, Z.; Vécsei, L. Current medical aspects of pantethine. Ideggyogy Szle. 2009, 62, 220–229.
- Ono, S.; Kameda, K.; Abiko, Y. Metabolism of panthethine in the rat. J. Nutr. Sci. Vitaminol. 1974, 20, 203–213.
- Reichlin, S.; Bollinger-Gruber, J.A. Pantethine, a cysteamine precursor, depletes immunoreactive somatostatin and prolactin in the rat. Endocrinology 1985, 112, 492–495.
- Jeitner, T.M.; Oliver, J.R. The depletion of plasma prolactin by pantethine in oestrogen primed hyperprolactinaemic rats. J. Endocrinol. 1990, 124, 397–402.
- Vécsei, L.; Widerlov, E.; Alling, C. Effects of pantethine, cysteamine and pantothenic acid on open-field behavior and brain catecholamines in rats. Arch. Int. Pharmacodyn. Ther. 1989, 300, 14–21.
- Nagiel-Ostaszewski, I.; Lau-Cam, C.A. Protection by pantethine, pantothenic acid and cystamine against carbontetrachloride-induced hepatotoxicity in the rat. Res. Commun. Chem. Pathol. Pharmacol. 1990, 67, 289–292.
- Nebbioso, M.; Pranno, F.; Pescosolido, N. Lipoic acid in animal models and clinical use in diabetic retinopathy. Expert Opin. Pharmacother. 2013, 14, 1829–1838.
- Reed, L.J. Metabolic functions of thiamine and lipoic acid. Physiol. Rev. 1953, 33, 544–559.
- Packer, L.; Witt, E.H.; Tritschler, H.J. Alpha-Lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 1995, 19, 227–250.
- Smith, A.R.; Shenvi, S.V.; Widlansky, M.; Suh, J.H.; Hagen, T.M. Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr. Med. Chem. 2004, 11, 1135–1146.
- Biewenga, G.P.; Haenen, G.R.; Bast, A. The pharmacology of the antioxidant lipoic acid. Gen. Pharmacol. Vasc. Syst. 1997, 29, 315–331.
- Packer, L.; Tritschler, H.J.; Wessel, K. Neuroprotection by the metabolic antioxidant alphalipoic acid. Free Radic. Biol. Med. 1997, 22, 359–378.
- Moini, H.; Packer, L.; Saris, N.-E.L. Antioxidant and Prooxidant Activities of α-Lipoic Acid and Dihydrolipoic Acid. Toxicol. Appl. Pharmacol. 2002, 182, 84–90.
- Kagan, V.E.; Shvedova, A.; Serbinova, E.; Khan, S.; Swanson, C.; Powell, R.; Packer, L. Dihydrolipoic acid-a universal antioxidant both in the membrane and in the aqueous phase. Biochem. Pharmacol. 1992, 44, 1637–1649.
- Moura, F.A.; de Andrade, K.Q.; dos Santos, J.C.; Goulart, M.O. Lipoic Acid: Its Antioxidant and Anti-Inflammatory Role and Clinical Applications. Curr. Top. Med. Chem. 2015, 15, 458–483.
- Yan, W.; Li, N.; Hu, X.; Huang, Y.; Zhang, W.; Wang, Q.; Wang, F.; Wang, C.; Zhai, X.; Xu, R.; et al. Effect of oral ALA supplementation on oxidative stress and insulin sensitivity among overweight/obese adults: A double-blinded, randomized, controlled, cross-over intervention trial. Int. J. Cardiol. 2013, 167, 602–603.
- Kim, M.-S.; Park, J.-Y.; Namkoong, C.; Jang, P.-G.; Ryu, J.-W.; Song, H.-S.; Yun, J.-Y.; Namgoong, I.-S.; Ha, J.; Park, I.-S.; et al. Anti-obesity effects of alpha-lipoic acid mediated by suppression of hypothalamic AMP-activated protein kinase. Nat. Med. 2004, 10, 727–733.
- Thakurta, I.G.; Chattopadhyay, M.; Ghosh, A.; Chakrabarti, S. Dietary supplementation with N-acetyl cysteine, α-tocopherol and α-lipoic acid reduces the extent of oxidative stress and proinflammatory state in aged rat brain. Biogerontology 2012, 13, 479–488.
- Al Abdan, M. Alfa-lipoic acid controls tumor growth and modulates hepatic redox state in Ehrlich-ascites-carcinoma-bearing mice. Sci. World J. 2012, 2012, 509838.
- Martins, V.D.; Manfredini, V.; Peralba, M.C.R.; Benfato, M.S. Alpha-lipoic acid modifies oxidative stress parameters in sickle cell trait subjects and sickle cell patients. Clin. Nutr. 2009, 28, 192–197.
- Bae, S.C.; Jung, W.J.; Lee, E.J.; Yu, R.; Sung, M.K. Effects of antioxidant supplements intervention on the level of plasma inflammatory molecules and disease severity of rheumatoid arthritis patients. J. Am. Coll. Nutr. 2009, 28, 56–62.
- Mauro, G.L.; Cataldo, P.; Barbera, G.; Sanfilippo, A. α-Lipoic acid and superoxide dismutase in the management of chronic neck pain: A prospective randomized study. Drugs R D 2014, 14, 1–7.
- Gu, X.; Zhang, S.; Wu, J.; Tang, Z.; Lu, Z.; Li, H.; Liu, C.; Chen, L.; Ning, G. Efficacy and safety of high-dose α-lipoic acid in the treatment of diabetic polyneuropathy. Zhonghua Yi Xue Za Zhi 2010, 90, 2473–2476.
- Ali, A.M.; Awad, T.G.; Al-Adl, N.M. Efficacy of combined topiramate/thioctic acid therapy in migraine prophylaxis. Saudi Pharm. J. 2010, 18, 239–243.
- Trivedi, P.P.; Jena, G.B. Role of α-lipoic acid in dextran sulfate sodium-induced ulcerative colitis in mice: Studies on inflammation, oxidative stress, DNA damage and fibrosis. Food Chem. Toxicol. 2013, 59, 339–355.
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta 2009, 1790, 1149–1160.


