Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eva Maria Verdugo-Sivianes | + 4458 word(s) | 4458 | 2021-05-26 06:13:23 | | | |
| 2 | Peter Tang | Meta information modification | 4458 | 2021-05-27 03:36:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Verdugo-Sivianes, E. Holoenzyme PP1-SPN in Cell Cycle. Encyclopedia. Available online: https://encyclopedia.pub/entry/10135 (accessed on 07 February 2026).
Verdugo-Sivianes E. Holoenzyme PP1-SPN in Cell Cycle. Encyclopedia. Available at: https://encyclopedia.pub/entry/10135. Accessed February 07, 2026.
Verdugo-Sivianes, Eva. "Holoenzyme PP1-SPN in Cell Cycle" Encyclopedia, https://encyclopedia.pub/entry/10135 (accessed February 07, 2026).
Verdugo-Sivianes, E. (2021, May 26). Holoenzyme PP1-SPN in Cell Cycle. In Encyclopedia. https://encyclopedia.pub/entry/10135
Verdugo-Sivianes, Eva. "Holoenzyme PP1-SPN in Cell Cycle." Encyclopedia. Web. 26 May, 2021.
Copy Citation
Cell cycle progression is highly regulated by modulating the phosphorylation status of retinoblastoma (RB) family proteins. This process is controlled by a balance in the action of kinases, such as the complexes formed by cyclin-dependent kinases (CDKs) and cyclins, and phosphatases, mainly the protein phosphatase 1 (PP1). The PP1-Spinophilin (SPN) holoenzyme has been described as the main phosphatase responsible for the dephosphorylation of RB proteins during the G0/G1 transition and at the end of G1.
RB family proteins
pocket proteins
cell cycle
phosphatase PP1
spinophilin
PPP1R9B
cancer
tumorigenesis
1. Introduction
During tumor development, cells undergo a series of genetic and/or epigenetic alterations, which gives them selective advantages over the environment, generating cancer cells. Many processes are involved in tumorigenesis, and one of the most important is the deregulation of the cell cycle. Therefore, cancer cells have undergone mutations deregulating the cell cycle that make them grow uncontrollably [1][2].
Cell cycle progression from one phase of the cycle to another is highly regulated through protein phosphorylation. In the G1 phase, there is a special checkpoint called restriction or R point at which the cell decides if it is ready to enter the cell cycle. The R point is controlled by the phosphorylation status of the retinoblastoma protein (pRB), a tumor suppressor protein whose main function is to inhibit cell cycle progression in G1 by binding E2F transcription factors and, thus, repressing E2F-target genes necessary to advance the cell cycle. Thus, until the pRB is phosphorylated and inhibited, cells cannot pass the R point and enter the cell cycle. Therefore, the G1/S transition is one of the most important checkpoints in the cell cycle [3][4][5][6][7][8]. Phosphorylation of the pRB inhibits its cell cycle restraining function by releasing E2F transcription factors. This phosphorylation is catalyzed by the complexes formed by cyclin-dependent kinases (CDKs) and cyclins. CDKs are serine-threonine kinases regulated by cyclins, proteins with cyclical expression whose levels increase and decrease drastically throughout the cell cycle, periodically activating CDKs [4][5][7][8][9][10][11][12][13][14][15][16][17]. The activity of CDKs/cyclins complexes can also be inhibited by the action of cyclin-dependent inhibitors, which can be divided into two families: the CIP/KIP family, composed of p21CIP1, p27KIP1 and p57KIP2, which inhibit most of CDKs/cyclin complexes; and the INK4 family, formed by p16INK4A, p15INK4B, p18INK4C and p19INK4D, which inhibit G1 CDKs, especially CDK4 and CDK6 [4][5][8][15][17][18]. However, the dephosphorylation of the pRB to restore the cell cycle, which is mainly mediated by the protein phosphatase 1 (PP1), is also very important and much more overlooked.
In this review, we describe the regulation of the phosphorylation status of the pRB and the other members of the RB family of tumor suppressors to emphasize not only their inactivation by phosphorylation but also their dephosphorylation to restore the cell cycle, two mechanisms that are frequently altered during tumorigenesis.
2. RB Family Proteins
The proteins of the pRB family or RB proteins are pRB itself, p107 (RBL1) and p130 (RBL2), three very similar proteins that share some biochemical properties and some functions [7][19][20][21]. These proteins are also known as pocket proteins because they have a domain -the pocket- capable of binding to different proteins and transcription factors. This pocket is composed of a smaller pocket, with two well-organized subdomains (A and B) separated by a less structured spacer region and the C-terminal region [4][17][18][22][23][24][25]. Specifically, the A/B subdomains bind to proteins containing the LXCXE motif, where “X” could be any amino acid. The PP1 phosphatase presents a variant of the motif (LXSXE) capable of binding to the pRB, while the E2F factors do not present this motif since their binding requires the entire pocket, including the C-terminal domain (Figure 1a) [4][7][17][18][22][26][27]. In addition, the pRB presents in this C-terminal region a special coupling site for E2F1 and a binding region for CDK2/cyclin A, CDK2/cyclin E, and PP1, while other CDKs bind to the N-terminus [7]. At least 16 phosphorylation residues are present in the pRB, all serine and threonine [15][17][28]. Phosphorylation of the pRB breaks the binding with different proteins; however, no kinase is capable of phosphorylating all of the pRB residues at the same time. The complete inactivation of the pRB requires sequential phosphorylation by different CDK/cyclin complexes, and depending on the residues that are phosphorylated, different proteins will dissociate sequentially, regulating the cycle-dependent genes differentially [4][18][26]. The structures of p107 and p130 are very similar to that of the pRB, but they are more related to each other (~50%) than to the pRB (20–30%). This is because their spacer region is larger and both present an insertion in the B subdomain of the small pocket and a region of homology at the N-terminus that allows them to act as inhibitors of CDKs (Figure 1a) [19][20][29][30][31][32][33]. Therefore, the pocket domain allows the association of RB proteins with many different proteins and transcription factors, some of them common and others specific to each protein so that, although there is not complete redundancy, there are some compensation mechanisms [20][21][23][27][34].
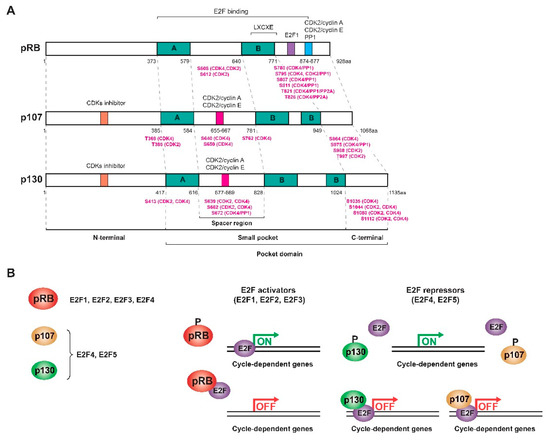
Figure 1. RB family proteins or pocket proteins. (A) Scheme of the structure of the three proteins of the RB family showing the different domains and motifs and the most relevant phosphorylated residues (highlighted in pink) with their respective kinases/phosphatases. (B) Interaction and regulation mechanisms of the pocket proteins with the different E2F transcription factors. Figure adapted from [19][20][21][35].
This family of proteins constitutes one of the major regulators of the cell cycle. They act by inhibiting transactivation mediated by activating E2F factors as well as forming complexes with E2F repressor factors to repress transcription and inhibit G1/S transition [36]. E2F levels vary throughout the cycle: while E2F1, E2F2, and E2F3 levels increase during the G1/S transition to induce proliferation, E2F4 and E2F5 are mostly expressed in resting cells [19]. However, the subset of E2F-dependent genes that each protein in the RB family regulates is different since each one interacts with different E2F factors: the pRB sequesters the activating factors E2F1-4, while p107 and p130 bind to repressor factors E2F4-5, although in the absence of E2F4, factors E2F1 and E2F3 can bind to p107 and p130 to compensate for their function [5][18][19][20][32][33][37][38]. E2F4 and E2F5 factors are expressed throughout the cycle, but during G0/G1, they bind p130 and p107 in the nucleus to form a repressor complex. In turn, the pRB sequesters E2F1, E2F2, and E2F3 to prevent binding to the corresponding promoters. At the end of G1, all the pocket proteins are phosphorylated and dissociate from the E2F factors so that E2F4-5 translocate to the cytoplasm and E2F1-3 bind to different promoters (Figure 1b) [36]. p130 (or p107) also mediates the repression of cell cycle genes as part of the DREAM (dimerization partner (DP), RB-like, E2F and multi-vulval class B (MuvB)) complex during quiescence [39]. Therefore, like the pRB, the activity of p107 and p130 is regulated during the cell cycle by controlling their state of phosphorylation in serine and threonine residues by the action of CDK/cyclin complexes in the middle/end of G1. When these proteins are dephosphorylated, they act as transcriptional repressors, while when they are phosphorylated, they are inactivated and dissociate from the E2Fs, allowing the transcription of genes involved in the cell cycle [19][20][21][36].
RB family proteins bind to numerous proteins, so they have other cellular functions beyond the control of the cell cycle. More than 200 proteins that interact with the pRB have been described, such as E2F transcription factors, cyclin D, MDM2, p53, and PP1, as well as other transcription factors and proteins related to differentiation, cell lineage identity, stemness invasion, apoptosis, senescence, angiogenesis, immune response and metabolism [4][17][18][22][23][28][40][41][42][43]. The pRB is an important tumor suppressor, and it is frequently inactivated, directly or indirectly, in many human tumors, promoting tumorigenesis [3][4][5][6][7][8][41][42][44]. Although p107 is hardly mutated in human tumors, and mice with mutations in p107 do not develop spontaneous tumors [30], the overexpression of hypophosphorylated p107 can induce G1 to stop in some cell types [32][37]. p130 is also not frequently mutated in cancer, but its levels are extremely low in some tumors due to its role in quiescence and differentiation [19]. Therefore, beyond genetic redundancy, the pRB could have some tumor suppressor functions that are not shared with p107 and p130 [5][7][20][23][34][35][42][45][46].
3. Protein Phosphatase 1 (PP1)
Protein phosphorylation is the major mechanism for regulating cellular functions since more than 70% of eukaryotic proteins are regulated by phosphorylation, mainly at serine and threonine residues [47][48]. This process is controlled by a balance in the action of kinases and phosphatases; however, while there are more than 400 genes that code for serine-threonine kinases, there are fewer than 40 genes that code for serine-threonine phosphatases. This is because phosphatases are enzymes formed by a catalytic subunit capable of interacting with numerous regulatory proteins to form different complexes (holoenzymes) with different locations and substrate specificities [4][12][47][48]. The largest subfamily of serine-threonine phosphatases is the phosphoprotein phosphatases (PPP), responsible for 95% of the phosphatase activity in cells. The most studied are PP1, PP2A and PP2B, but one-third of the dephosphorylation events in eukaryotic cells are only carried out by PP1 [4][12][47][49][50][51][52][53][54][55].
PP1 is an enzyme involved in the regulation of many cellular processes, such as protein synthesis, transcription, apoptosis, and cell cycle progression [47][51][55][56]. In mammals, three genes (PPP1CA, PPP1CB, and PPP1CC) encode four isoforms of the catalytic subunit of PP1: PP1α, PP1β, PP1γ1, and PP1γ2. These four isoforms are expressed in all tissues and compartments, although PP1γ2 is only expressed in testes [10][47][48][54][57][58][59][60][61]. In addition, all isoforms are found in the nucleus, but PP1β and PP1γ present a special accumulation in the nucleolus [47]. The sequence of the three isoforms is highly conserved: 93% between PP1γ1 and PP1γ2 and 85% between PP1β and PP1γ2, although the N-terminal and the C-terminal present greater differences [10][47][48][54][57][58][59][60].
PP1 is involved in a large number of functions; however, it does not have substrate specificity by itself, and it needs to interact with multiple regulatory proteins. Thus, the catalytic subunit of PP1 (PPP1C) can bind and form different holoenzymes with multiple regulatory proteins (PPP1R), also known as regulatory interactors of protein phosphatase 1 (RIPPOs). RIPPOs direct PP1 towards specific substrates to perform specific functions, preventing the dephosphorylation of substrates by occupying the PP1 binding site or promoting the dephosphorylation by directing PP1 to specific cell locations. Additionally, some RIPPOs can be PP1 substrates [47][48][54][61][62][63][64][65]. Currently, approximately 200 regulatory proteins of PP1 are known, and most of them do not show similarity in their sequence [47][52][54][55][64]. Approximately 90% of PP1-interacting proteins bind to it through the PP1 binding motif RVxF, which generally consists of the consensus sequence: (K/R) (R/K) (V/I) (x) (F/W) where “x” can be any residue except F, I, M, Y, D or P [48][56][62][64]. However, this interaction is unique to each regulatory protein, so that mutations in the RVxF motif would prevent the binding of the regulatory protein to PP1 but would not affect the binding of the substrate or the formation of other holoenzymes [56]. In addition to the RVxF motif, there are other binding motifs that not only stabilize the binding to PP1 but also modulate its activity and specificity [47][48][54][62][63][64].
PP1 is the protein phosphatase responsible for dephosphorylating and activating the pRB from the exit of mitosis to the middle of G1 [3][12][18][35][57][66][67][68]. The interaction between the pRB and PP1 occurs through the PP1 binding motif, with a high affinity and is direct, without the need for regulatory proteins. However, these proteins serve as regulators of the pRB-specific PP1 activity and are context/tissue-dependent, since PP1 alone cannot regulate the pRB [21][35][60]. PP1 forms a complex with both the hypophosphorylated and hyperphosphorylated pRB; however, if the pRB is phosphorylated at certain residues, it cannot bind PP1, so PP1 needs the help of a regulatory protein to gain access to the pRB to dephosphorylate it. Indeed, the preferred sites for PP1 to dephosphorylate the pRB are T356 and S807/811. After dephosphorylation, PP1 remains bound to the hypophosphorylated pRB forming a complex that lasts until entry into G1, preventing its phosphorylation [4][14][57][66][67][69]. In addition, PP1 and the CDK/cyclin complexes share the binding site to the pRB, so there is a competition for the substrate between the kinase and the phosphatase activities [60]. On the other hand, p107 and p130 also present the PP1 binding motif. The interaction between these two proteins and PP1 has been described in double hybrid assays and by co-immunoprecipitation [60][70]. However, this interaction seems to be much weaker and lasts less time than that between the pRB and PP1 [21][71].
The regulation of PP1 is complex and is controlled by both phosphorylation by different kinases and the action of inhibitors and regulatory proteins that modulate its substrate specificity and activity [3][4][5][57][61][69][72][73]. On the one hand, PP1 is phosphorylated and inactivated during the cell cycle by CDK2/cyclin E, CDK2/cyclin A, CDK1/cyclin A and CDK1/cyclin B to avoid pRB dephosphorylation. Specifically, the phosphorylation of PP1 at the T320 residue plays an important role in the G1/S transition and during mitosis [3][61][74][75]. On the other hand, CDK1/cyclin B phosphorylates and inactivates PP1 with the help of the PP1 Inhibitor-2 (I2) during the onset and the middle of mitosis [4][76]. At the exit of mitosis, PP1 is activated by destroying the CDK1/cyclin B complex and by PP1 autodephosphorylation at residue T320 [14][18][47][66][74][76], which is inhibited during mitosis by the binding of Inhibitor-1 (I1) to PP1 [47][76].
The three isoforms of the catalytic subunit of PP1 bind the pRB similarly since the interaction region is conserved. In addition, all of them have the ability to dephosphorylate the pRB but present different activities in the different phases of the cycle [3][21][60][77]. During G1 and in the G1/S transition, PP1α is the main isoform that controls the pRB [51][69]. When cells enter mitosis, all isoforms are phosphorylated and inactivated since there is an increase in the phosphorylation of the PP1α protein at serine residues and PP1β and PP1γ1 proteins at threonine residues. Finally, at the end of the mitosis phase, PP1α and PP1β activity increases while PP1γ1 remains phosphorylated and with low activity [72][73]. Indeed, PP1β is the most active isoform during mitosis, but its activity does not persist during G1 [18][21][60][72][73][77]. Therefore, the dephosphorylation of the pRB is regulated in a sequential and temporal manner, and the three isoforms of the catalytic subunit of PP1 bind to different regulatory proteins to form different holoenzymes with different preferences for phosphorylation sites, similar to pRB phosphorylation by CDK/cyclin complexes [5][18][21][77]. In fact, different holoenzymes of PP1 may form during the cell cycle to control the dephosphorylation of the pRB since the three isoforms of the catalytic subunit of PP1 present different activities in the different phases of the cell cycle [3][4][5][57][69][72].
In this regard, the study of PP1 regulatory proteins involved in the cell cycle is essential since mutations in the catalytic subunit of PP1 or in the regulatory proteins that prevent binding to the pRB will promote phosphorylation of the pRB and, eventually, cell transformation [4][14][66][67][69].
4. SPN, a PP1 Regulatory Protein
Spinophilin (SPN), also known as PPP1R9B and NEURABIN-2, is a PP1 regulatory protein widely expressed in many tissues such as the brain, lung, testes, colon, breast, among others [78][79][80][81][82][83][84][85][86][87][88]. SPN presents in its structure a PP1 binding domain, but the SPN-PP1 interaction occurs not only through the RVxF motif but also by forming multiple interactions with different regions of PP1, including part of the C-terminus of PP1 [62][79][81][82][89][90]. Indeed, the structure of SPN suggests that it is a multifunctional protein that functions as a scaffold protein by recruiting many different proteins into different cell signaling pathways and promoting protein-protein interaction [79][81][82][89]. SPN has been shown to be located in the cytoplasm and in the plasma membrane of cells but could also be expressed in the nucleus [82][91]. We recently demonstrated that SPN co-localizes with PP1α and PP1γ in the nucleus and in the cytoplasm of cells [70].
One of the main functions of SPN is to help PP1 dephosphorylate the pRB [28][70][84]. SPN interacts with PP1α and PP1γ but not with PP1β [59][70]. In addition, SPN interacts specifically with both total and phosphorylated pRB (P-pRB) in Ser807/811, two of the preferred PP1 dephosphorylation sites [10][70]. SPN is also able to bind and dephosphorylate phosphorylated p107 (P-p107) in Ser975, a homologous residue to Ser807/811 in P-pRB, and phosphorylated p130 (P-p130) in Ser672, an important residue implicated in the stability of p130 during the cell cycle and a possible dephosphorylation site of PP1 [20][34][92][93][70]. Therefore, the PP1-SPN holoenzyme is not exclusive to the pRB but acts over all the pocket family proteins, although another phosphatase could dephosphorylate P-p107 and P-p130 in other contexts [70].
Cell cycle assays, in which cells were synchronized at G0 through serum deprivation or at the end of G1 after mimosine treatment, demonstrated that the PP1-SPN holoenzyme regulates the dephosphorylation of pocket proteins during the G0/G1 transition and at the end of G1. However, cell cycle assays in which cells were synchronized at the G2/M transition after nocodazole treatment showed that this holoenzyme does not act during mitosis when PP1β is the most active isoform and does not interact with SPN. Therefore, the PP1-SPN holoenzyme is formed by SPN and either PP1α or PP1γ and is involved in the dephosphorylation of pocket proteins exclusively during the G0/G1 transition and at the end of G1, but not during the G2/M transition or the mitosis phase [70]. Instead, PP1β could bind to a different PP1 regulatory protein at the exit of mitosis, but this other regulatory protein remains unidentified (Figure 2a) [4][14][57]. It has been reported that phosphatase nuclear targeting subunit (PNUTS) is a PP1 inhibitory protein with an important role in controlling PP1 activity during mitosis by inhibiting pRB dephosphorylation. However, PNUTS is only associated with a small proportion of PP1, and other proteins beyond PNUTS and SPN must regulate PP1 during the cell cycle [70][94]. PNUTS is a context-dependent PP1 regulatory protein, and the role of SPN in PP1 regulation and pocket protein dephosphorylation might also be dependent on the context, regarding either the cell cycle phase or the subcellular localization [70]. In addition, the pRB could function as a substrate or as a regulatory protein for PP1 since different subpopulations of the pRB perform different functions depending on the type of phosphorylation [67]. Different holoenzymes could be involved in the sequential control of pocket protein dephosphorylation during cell cycle progression, and each holoenzyme might have a distinct specificity for different phosphorylated residues, similar to CDK/cyclin complexes; therefore, initial dephosphorylation would be necessary to induce a conformational change before any other holoenzyme gains access to the different residues. In addition, whether the dephosphorylation of pocket proteins by PP1 in mitosis and in G1 occurs through a single mechanism or if different substrates are recognized by different holoenzymes must be determined [18][70].
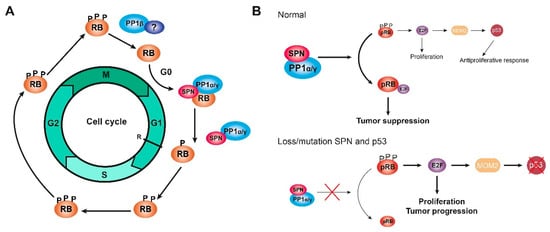
Figure 2. The holoenzyme PP1-SPN. (A) Scheme of the dephosphorylation of RB proteins (named here RB to refer to the three pocket proteins at the same time) during G1 by the holoenzyme PP1α/γ-SPN. The dephosphorylation of RB proteins might be regulated by PP1β in complex with an unknown regulatory protein during the end of mitosis. (B) Scheme of the mechanism of the holoenzyme PP1-SPN in tumorigenesis, comparing a normal situation (upper) and a tumoral situation in which SPN is lost/mutated, and p53 is mutated (bottom). Only when p53 is mutated, the loss/mutation of SPN can induce cell proliferation and tumor progression, evading the neutralizing response of p53. Figure adapted from [70][80][81].
On the other hand, SPN is phosphorylated by different protein kinases: protein kinase A (PKA) phosphorylates SPN in S97 and S177, calcium/calmodulin-dependent protein kinase II (CaMKII) phosphorylates SPN in S100 and S116, cyclin-dependent protein kinase-5 (CDK5) in S17 and mitogen-activated protein kinase-1 (MAPK1 or ERK2) in S15 and S205 [82][95][96][97], some of them also phosphorylate PP1. Indeed, phosphorylation of PP1 at Thr311/320 by CDK5 enhances its association with SPN, but phosphorylation of SPN by CDK5 at Ser17 is not responsible for the increased interaction between PP1 and SPN [98]. Although this enhanced interaction induced by CDK5 could add another layer of complexity to the regulation of RB proteins dephosphorylation during the cell cycle, it should be studied in depth to extract any conclusion.
In addition, SPN forms a different complex with PP1 and DCX, a microtubule-associated protein that binds to tubulin and actin. DCX is phosphorylated by CDK5, which prevents its binding to microtubules, and dephosphorylated by PP1 through interaction with SPN so that this axis regulates the maintenance of microtubules [99][100][101].
Therefore, the holoenzyme PP1-SPN performs different functions in the cell both in the nucleus and in the cytoplasm, depending on the association with different proteins.
5. SPN as a Tumor Suppressor Dependent on PP1 and pRB
The locus of SPN is located on chromosome 17 at the 17q21.33 position, a chromosomal region frequently associated with microsatellite instability and loss of heterozygosity and a high density of well-known tumor suppressor genes such as BRCA1. This loss of heterozygosity in the 17q21 region has been reported in different tumors, such as breast, ovarian, lung, prostate, colorectal, gastric, renal, and lung cancer [81][102][103][104][105][106][107][108]. Several studies suggested the existence of a new tumor suppressor gene located in the 17q21 region, and eventually, SPN was identified as this new gene [102]. Currently, SPN has been described as a tumor suppressor gene in the context of different human tumors, such as renal carcinomas, lung adenocarcinomas, ovarian carcinoma, chronic myeloid leukemia, gastric and colorectal cancer, head and neck carcinoma, hepatocellular carcinoma, and breast cancer [81][84][85][86][109][110].
Various studies in lung cancer have corroborated that SPN has a prognostic and predictive value in this type of tumor since the downregulation of this gene together with p53 mutations are associated with worse survival. In addition, a correlation between the decrease in SPN levels and low levels of the three catalytic subunits of PP1 was observed, and this combination was associated with a worse prognosis in squamous cell carcinoma. Loss of SPN has also shown a decrease in PP1 expression and activity in the brain tissue of SPN-knockout mice [111][112]. The SPN/PPP1C ratio could serve as a response biomarker due to its prognostic and predictive value in lung cancer. Indeed, a direct correlation was observed between the SPN/PPP1C ratio and the response to different drugs commonly used in the clinic, such as oxaliplatin and bortezomib; therefore, the SPN/PPP1C ratio could also be used as a therapy response marker in those types of tumors [87].
In breast cancer, SPN plays an important role as a tumor suppressor. In vivo studies using Spn-knockout mice reported that the absence of SPN decreased the life expectancy of mice and increased the number of spontaneous tumors such as lymphomas. Spn−/− mice also presented an increased cell proliferation of certain tissues, such as the breast ducts, and both Spn+/− and Spn−/− mice showed more ramifications in this tissue. Indeed, Spn−/− mice did not express SPN in the mammary ducts [83][113]. Mouse embryonic fibroblasts generated from Spn−/− mice showed lower levels of PP1α and decreased PP1 activity, which in turn produced higher levels of phosphorylated pRB and increased p53 activity [80]. Additionally, the combination of the loss of SPN and p53 using p53-knockout mice induced preneoplastic lesions in the mammary glands, suggesting that the loss of SPN increases the p53 response similarly to oncogene-induced senescence. Thus, once spontaneous tumors appear, and p53 is lost, the loss of SPN increases their aggressiveness [81][83].
The loss of SPN has been reported in 15% of breast tumors, correlating with a higher histological grade, a less differentiated phenotype, and worse survival. In fact, both SPN and p53 are lost in triple-negative tumors, and this combination makes tumors more aggressive [114]. The downregulation of SPN in breast cancer cell lines increases some tumorigenic properties of the cells, such as the ability to proliferate or to form colonies, and some cancer stem cell properties, such as the formation of tumorspheres and the expression of stem cell genes. This effect depends on PP1 activity since the downregulation of PP1α mimics the effect of the downregulation of SPN [84][88]. Therefore, in tumor cells, the loss of SPN induces a proliferative response by reducing PP1α levels and increasing hyperphosphorylated and inactive pRB levels, which in turn activate p53 and neutralize the proliferative response. However, the loss or mutation of SPN is frequently associated with p53 mutations; therefore, in the absence of p53, the loss or mutation of SPN levels produces an increase in cell proliferation, and the tumorigenic properties of the cells are enhanced (Figure 2b) [70][80][81].
In addition, the downregulation of SPN also induces an increase in the stemness properties of the cells, such as the expression of some cancer stem cell markers (NANOG, OCT4, SOX2, and KLF4) and enrichment in CD44+/CD24- cells, cancer-initiating cells in breast tumors with stem cell properties [84][115]. Therefore, the loss of SPN in breast cancer induces an increase in the cancer stem cell pool, which worsens the response of those tumors to chemotherapy [70][84][88][116].
On the other hand, thirty-nine mutations in the region of interaction between SPN and PP1 have been identified [70]. The mutation of SPN, SPN-A566V, has an oncogenic effect since the expression of this mutation in breast cancer cell lines induces an increase in the tumorigenic and stemness properties of the cells depending on p53 mutations [70]. SPN-A566V affects the PP1 phosphatase activity of the holoenzyme, especially over the pocket proteins. Indeed, SPN-A566V did not interrupt the SPN-pRB interaction but decreased the capacity of the holoenzyme PP1-SPN to dephosphorylate P-pRB. The mutation of SPN affects the interaction between SPN and p107 and p130, and decrease the capacity of the holoenzyme PP1-SPN to dephosphorylate them [70]. Cells that overexpress SPN-A566V have high levels of the P-pRB, P-p107, and partially P-p130 during the G0/G1 transition and at the end of G1, which could mean that they have a shorter G1 phase in order to proliferate more rapidly. This mutation also induced an increase in the cancer stem cell pool and the expression of NANOG, OCT4, and SOX2 [70]. Recently, the pRB was reported to be directly involved in the transcriptional regulation of the pluripotency genes OCT4 and SOX2 [117]. When the pRB is dephosphorylated and active, the OCT4 and SOX2 promoters are inhibited [118]; thus, the P-pRB may promote OCT4/SOX2 expression in SPN-A566V cells, which in turn induces NANOG [119][120]. At the same time, OCT4 regulates the self-renewal and differentiation of embryonic stem cells and controls the cell cycle by increasing CDK/cyclin levels during the G1 phase and by preventing pRB dephosphorylation by PP1 [118][121][122]. Therefore, a connection between the cell cycle and stem cell biology was also proposed via SPN/PP1/pocket proteins, but further studies are needed to clarify whether the PP1-SPN holoenzyme plays any role in the OCT4/pRB self-regulatory circuit [70].
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Berndt, N.; Dohadwala, M.; Liu, C.W. Constitutively active protein phosphatase 1alpha causes Rb-dependent G1 arrest in human cancer cells. Curr. Biol. 1997, 7, 375–386.
- Rubin, E.; Tamrakar, S.; Ludlow, J.W. Protein phosphatase type 1, the product of the retinoblastoma susceptibility gene, and cell cycle control. Front. Biosci. 1998, 3, D1209–D1219.
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330.
- Planas-Silva, M.D.; Weinberg, R.A. The restriction point and control of cell proliferation. Curr. Opin. Cell Biol. 1997, 9, 768–772.
- Bartek, J.; Bartkova, J.; Lukas, J. The retinoblastoma protein pathway and the restriction point. Curr. Opin. Cell Biol. 1996, 8, 805–814.
- Schafer, K.A. The Cell Cycle: A Review. Vet. Pathol. 1998, 35, 461–478.
- Krtolica, A.; Krucher, N.A.; Ludlow, J.W. Molecular analysis of selected cell cycle regulatory proteins during aerobic and hypoxic maintenance of human ovarian carcinoma cells. Br. J. Cancer 1999, 80, 1875–1883.
- Berndt, N. Protein dephosphorylation and the intracellular control of the cell number. Front. Biosci. 1999, 4, D22–D42.
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149.
- Ludlow, J.W.; Nelson, D.A. Control and activity of type-1 serine/threonine protein phosphatase during the cell cycle. Semin. Cancer Biol. 1995, 6, 195–202.
- Kitagawa, M.; Higashi, H.; Jung, H.K.; Suzuki-Takahashi, I.; Ikeda, M.; Tamai, K.; Kato, J.Y.; Segawa, K.; Yoshida, E.; Nishimura, S.; et al. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 1996, 15, 7060–7069.
- Nelson, D.A.; Ludlow, J.W. Characterization of the mitotic phase pRb-directed protein phosphatase activity. Oncogene 1997, 14, 2407–2415.
- Lundberg, A.S.; Weinberg, R.A. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell Biol. 1998, 18, 753–761.
- Lundberg, A.S.; Weinberg, R.A. Control of the cell cycle and apoptosis. Eur. J. Cancer 1999, 35, 1886–1894.
- Harbour, J.W.; Luo, R.X.; Dei Santi, A.; Postigo, A.A.; Dean, D.C. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98, 859–869.
- Tamrakar, S.; Rubin, E.; Ludlow, J.W. Role of pRB dephosphorylation in cell cycle regulation. Front. Biosci. 2000, 5, D121–D137.
- Classon, M.; Dyson, N. p107 and p130: Versatile proteins with interesting pockets. Exp. Cell Res. 2001, 264, 135–147.
- Graña, X.; Garriga, J.; Mayol, X. Role of the retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene 1998, 17, 3365–3383.
- Kolupaeva, V.; Janssens, V. PP1 and PP2A phosphatases—Cooperating partners in modulating retinoblastoma protein activation. FEBS J. 2013, 280, 627–643.
- Di Fiore, R.; D’Anneo, A.; Tesoriere, G.; Vento, R. RB1 in cancer: Different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J. Cell Physiol. 2013, 228, 1676–1687.
- Chinnam, M.; Goodrich, D.W. RB1, Development, and Cancer. Curr. Top. Dev. Biol. 2011, 94, 129–169.
- Guzman, F.; Fazeli, Y.; Khuu, M.; Salcido, K.; Singh, S.; Benavente, C.A. Retinoblastoma tumor suppressor protein roles in epigenetic regulation. Cancers 2020, 12, 2807.
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct. Target. Ther. 2020, 5, 90.
- Adams, P.D. Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochim. Biophys. Acta 2001, 1471, M123–M133.
- Genovese, C.; Trani, D.; Caputi, M.; Claudio, P.P. Cell cycle control and beyond: Emerging roles for the retinoblastoma gene family. Oncogene 2006, 25, 5201–5209.
- Mittnacht, S. The retinoblastoma protein—From bench to bedside. Eur. J. Cell Biol. 2005, 84, 97–107.
- Claudio, P.P.; Tonini, T.; Giordano, A. The retinoblastoma family: Twins or distant cousins? Genome Biol. 2002, 3, 1–9.
- Wirt, S.E.; Sage, J. p107 in the public eye: An Rb understudy and more. Cell Div. 2010, 5, 9.
- Garriga, J.; Limón, A.; Mayol, X.; Rane, S.G.; Albrecht, J.H.; Reddy, E.P.; Andrés, V.; Graña, X. Differential regulation of the retinoblastoma family of proteins during cell proliferation and differentiation. Biochem. J. 1998, 333, 645–654.
- Beijersbergen, R.L.; Carlée, L.; Kerkhoven, R.M.; Bernards, R. Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes Dev. 1995, 9, 1340–1353.
- Du, W.; Pogoriler, J. Retinoblastoma family genes. Oncogene 2006, 25, 5190–5200.
- Mayol, X.; Grana, X. The p130 pocket protein: Keeping order at cell cycle exit/re-entrance transitions. Front. Biosci. 1998, 3, d11–d24.
- Henley, S.A.; Dick, F.A. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012, 7, 10.
- Cobrinik, D. Pocket proteins and cell cycle control. Oncogene 2005, 24, 2796–2809.
- Xiao, Z.X.; Ginsberg, D.; Ewen, M.; Livingston, D.M. Regulation of the retinoblastoma protein-related protein p107 by G1 cyclin-associated kinases. Proc. Natl. Acad. Sci. USA 1996, 93, 4633–4637.
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262.
- Sadasivam, S.; DeCaprio, J.A. The DREAM complex: Master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer 2013, 13, 585–595.
- Liu, H.; Dibling, B.; Spike, B.; Dirlam, A.; Macleod, K. New roles for the RB tumor suppressor protein. Curr. Opin. Genet. Dev. 2004, 14, 55–64.
- Burkhart, D.L.; Morel, K.L.; Sheahan, A.V.; Richards, Z.A.; Ellis, L. The Role of RB in Prostate Cancer Progression. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; pp. 301–318.
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451.
- Classon, M.; Harlow, E. The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2002, 2, 910–917.
- Indovina, P.; Pentimalli, F.; Conti, D.; Giordano, A. Translating RB1 predictive value in clinical cancer therapy: Are we there yet? Biochem. Pharmacol. 2019, 16, 323–334.
- Mushtaq, M.; Gaza, H.V.; Kashuba, E.V. Role of the RB-Interacting Proteins in Stem Cell Biology. Adv. Cancer Res. 2016, 131, 133–157.
- Viatour, P.; Sage, J. Newly identified aspects of tumor suppression by RB. Dis. Model Mech. 2011, 4, 581–585.
- Rebelo, S.; Santos, M.; Martins, F.; da Cruz e Silva, E.F.; da Cruz e Silva, O.A.B. Protein phosphatase 1 is a key player in nuclear events. Cell Signal 2015, 27, 2589–2598.
- Peti, W.; Nairn, A.C.; Page, R. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 2013, 280, 596–611.
- Alberts, A.S.; Thorburn, A.M.; Shenolikar, S.; Mumby, M.C.; Feramisco, J.R. Regulation of cell cycle progression and nuclear affinity of the retinoblastoma protein by protein phosphatases. Proc. Natl. Acad. Sci. USA 1993, 90, 388–392.
- Walter, G.; Mumby, M. Protein serine/threonine phosphatases and cell transformation. Biochim. Biophys. Acta 1993, 1155, 207–226.
- Wera, S.; Hemmings, B.A. Serine/threonine protein phosphatases. Biochem. J. 1995, 311, 17–29.
- Cohen, P.T.W. Protein phosphatase 1—Targeted in many directions. J. Cell Sci. 2002, 115, 241–256.
- Brautigan, D.L. Flicking the switches: Phosphorylation of serine/threonine protein phosphatases. Semin. Cancer Biol. 1995, 6, 211–217.
- Dancheck, B.; Ragusa, M.J.; Allaire, M.; Nairn, A.C.; Peti, W. Molecular Investigations of the Structure and Function of the Protein Phosphatase 1:Spinophilin:Inhibitor-2 Heterotrimeric Complex. Biochemistry 2011, 50, 1238–1246.
- Cohen, P.T.; Brewis, N.D.; Hughes, V.; Mann, D.J. Protein serine/threonine phosphatases; an expanding family. FEBS Lett. 1990, 268, 355–359.
- Egloff, M.P.; Johnson, D.F.; Moorhead, G.; Cohen, P.T.; Cohen, P.; Barford, D. Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J. 1997, 16, 1876–1887.
- Nelson, D.A.; Krucher, N.A.; Ludlow, J.W. High molecular weight protein phosphatase type 1 dephosphorylates the retinoblastoma protein. J. Biol. Chem. 1997, 272, 4528–4535.
- Sasaki, K.; Shima, H.; Kitagawa, Y.; Irino, S.; Sugimura, T.; Nagao, M. Identification of Members of the Protein Phosphatase 1 Gene Family in the Rat and Enhanced Expression of Protein Phosphatase 1α Gene in Rat Hepatocellular Carcinomas. Jpn. J. Cancer Res. 1990, 81, 1272–1280.
- Terry-Lorenzo, R.T.; Carmody, L.C.; Voltz, J.W.; Connor, J.H.; Li, S.; Donelson Smith, F.; Milgram, S.L.; Colbran, R.J.; Shenolikar, S. The neuronal actin-binding proteins, neurabin I and neurabin II, recruit specific isoforms of protein phosphatase-1 catalytic subunits. J. Biol. Chem. 2002, 277, 27716–27724.
- Hirschi, A.; Cecchini, M.; Steinhardt, R.C.; Schamber, M.R.; Dick, F.A.; Rubin, S.M. An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nat. Struct. Mol. Biol. 2010, 17, 1051–1057.
- Felgueiras, J.; Jerónimo, C.; Fardilha, M. Protein phosphatase 1 in tumorigenesis: Is it worth a closer look? Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188433.
- Ragusa, M.J.; Dancheck, B.; Critton, D.A.; Nairn, A.C.; Page, R.; Peti, W. Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat. Struct. Mol. Biol. 2010, 17, 459–464.
- Ragusa, M.J.; Allaire, M.; Nairn, A.C.; Page, R.; Peti, W. Flexibility in the PP1:spinophilin holoenzyme. FEBS Lett. 2011, 585, 36–40.
- Heroes, E.; Lesage, B.; Görnemann, J.; Beullens, M.; Van Meervelt, L.; Bollen, M. The PP1 binding code: A molecular-lego strategy that governs specificity. FEBS J. 2013, 280, 584–595.
- Köhn, M. Turn and Face the Strange: § A New View on Phosphatases. ACS Cent. Sci. 2020, 6, 467–477.
- Ludlow, J.W.; Glendening, C.L.; Livingston, D.M.; DeCarprio, J.A. Specific enzymatic dephosphorylation of the retinoblastoma protein. Mol. Cell Biol. 1993, 13, 367–372.
- Tamrakar, S.; Mittnacht, S.; Ludlow, J.W. Binding of select forms of pRB to protein phosphatase type 1 independent of catalytic activity. Oncogene 1999, 18, 7803–7809.
- Durfee, T.; Becherer, K.; Chen, P.L.; Yeh, S.H.; Yang, Y.; Kilburn, A.E.; Lee, W.H.; Elledge, S.J. The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes Dev. 1993, 7, 555–569.
- Liu, C.W.; Wang, R.H.; Dohadwala, M.; Schönthal, A.H.; Villa-Moruzzi, E.; Berndt, N. Inhibitory phosphorylation of PP1alpha catalytic subunit during the G(1)/S transition. J. Biol. Chem. 1999, 274, 29470–29475.
- Verdugo-Sivianes, E.M.; Rojas, A.M.; Muñoz-Galván, S.; Otero-Albiol, D.; Carnero, A. Mutation of SPINOPHILIN (PPP1R9B) found in human tumors promotes the tumorigenic and stemness properties of cells. Theranostics 2021, 11, 3452–3471.
- Garriga, J.; Jayaraman, A.L.; Limón, A.; Jayadeva, G.; Sotillo, E.; Truongcao, M.; Patsialou, A.; Wadzinski, B.E.; Grana, X. A dynamic equilibrium between CDKs and PP2A modulates phosphorylation of pRB, p107 and p130. Cell Cycle 2004, 3, 1320–1330.
- Puntoni, F.; Villa-Moruzzi, E. Protein phosphatase-1 alpha, gamma 1, and delta: Changes in phosphorylation and activity in mitotic HeLa cells and in cells released from the mitotic block. Arch. Biochem. Biophys. 1997, 340, 177–184.
- Puntoni, F.; Villa-Moruzzi, E. Association of protein phosphatase-1delta with the retinoblastoma protein and reversible phosphatase activation in mitotic HeLa cells and in cells released from mitosis. Biochem. Biophys. Res. Commun. 1997, 235, 704–708.
- Dohadwala, M.; Silva, E.F.D.C.E.; Hall, F.L.; Williams, R.T.; Carbonaro-Hall, D.A.; Nairn, A.C.; Greengard, P.; Berndt, N. Phosphorylation and inactivation of protein phosphatase 1 by cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA 1994, 91, 6408–6412.
- Liu, C.W.Y.; Wang, R.-H.; Berndt, N. Protein phosphatase 1α activity prevents oncogenic transformation. Mol. Carcinog. 2006, 45, 648–656.
- Wu, J.Q.; Guo, J.Y.; Tang, W.; Yang, C.-S.; Freel, C.D.; Chen, C.; Nairn, A.C.; Kornbluth, S. PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat. Cell Biol. 2009, 11, 644–651.
- Rubin, E.; Mittnacht, S.; Villa-Moruzzi, E.; Ludlow, J.W. Site-specific and temporally-regulated retinoblastoma protein dephosphorylation by protein phosphatase type 1. Oncogene 2001, 20, 3776–3785.
- Allen, P.B.; Ouimet, C.C.; Greengard, P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc. Natl. Acad. Sci. USA 1997, 94, 9956–9961.
- Satoh, A.; Nakanishi, H.; Obaishi, H.; Wada, M.; Takahashi, K.; Satoh, K.; Hirao, K.; Nishioka, H.; Hata, Y.; Mizoguchi, A.; et al. Neurabin-II/Spinophilin. An Actin Filament-Binding Protein with One Pdz Domain Localized at Cadherin-Based Cell-Cell Adhesion Sites. J. Biol. Chem. 1998, 273, 3470–3475.
- Ferrer, I.; Blanco-Aparicio, C.; Peregrino, S.; Cañamero, M.; Fominaya, J.; Cecilia, Y.; Lleonart, M.; Hernandez-Losa, J.; Ramon y Cajal, S.; Carnero, A. Spinophilin acts as a tumor suppressor by regulating Rb phosphorylation. Cell Cycle 2011, 10, 2751–2762.
- Carnero, A. Spinophilin: A new tumor suppressor at 17q21. Curr. Mol. Med. 2012, 12, 528–535.
- Sarrouilhe, D.; di Tommaso, A.; Métayé, T.; Ladeveze, V. Spinophilin: From partners to functions. Biochimie 2006, 88, 1099–1113.
- Ferrer, I.; Peregrino, S.; Cañamero, M.; Cecilia, Y.; Blanco-Aparicio, C.; Carnero, A. Spinophilin loss contributes to tumorigenesis in vivo. Cell Cycle 2011, 10, 1948–1955.
- Ferrer, I.; Verdugo-Sivianes, E.M.; Castilla, M.A.; Melendez, R.; Marin, J.J.; Muňoz-Galvan, S.; Lopez-Guerra, J.L.; Vieites, B.; Ortiz-Gordillo, M.J.; De Leon, J.M.; et al. Loss of the tumor suppressor spinophilin (PPP1R9B) increases the cancer stem cell population in breast tumors. Oncogene 2016, 35, 2777–2788.
- Estevez-Garcia, P.; Lopez-Calderero, I.; Molina-Pinelo, S.; Muñoz-Galvan, S.; Salinas, A.; Gomez-Izquierdo, L.; Lucena-Cacace, A.; Felipe-Abrio, B.; Paz-Ares, L.; Garcia-Carbonero, R.; et al. Spinophilin loss correlates with poor patient prognosis in advanced stages of colon carcinoma. Clin. Cancer Res. 2013, 19, 3925–3935.
- Molina-Pinelo, S.; Ferrer, I.; Blanco-Aparicio, C.; Peregrino, S.; Pastor, M.D.; Alvarez-Vega, J.; Suarez, R.; Verge, M.; Marin, J.J.; Hernández-Losa, J.; et al. Down-regulation of spinophilin in lung tumours contributes to tumourigenesis. J. Pathol. 2011, 225, 73–82.
- Verdugo-Sivianes, E.M.; Navas, L.; Molina-Pinelo, S.; Ferrer, I.; Quintanal-Villalonga, A.; Peinado, J.; Garcia-Heredia, J.M.; Felipe-Abrio, B.; Muñoz-Galvan, S.; Marin, J.J.; et al. Coordinated downregulation of Spinophilin and the catalytic subunits of PP1, PPP1CA/B/C, contributes to a worse prognosis in lung cancer. Oncotarget 2017, 8, 105196–105210.
- Schwarzenbacher, D.; Stiegelbauer, V.; Deutsch, A.; Ress, A.L.; Aigelsreiter, A.; Schauer, S.; Wagner, K.; Langsenlehner, T.; Resel, M.; Gerger, A.; et al. Low spinophilin expression enhances aggressive biological behavior of breast cancer. Oncotarget 2015, 6, 11191–11202.
- Barnes, A.P.; Smith, F.D.; VanDongen, H.M.; VanDongen, A.M.J.; Milgram, S.L. The identification of a second actin-binding region in spinophilin/neurabin II. Mol. Brain Res. 2004, 124, 105–113.
- Carmody, L.C.; Baucum, A.J.; Bass, M.A.; Colbran, R.J. Selective targeting of the γ1 isoform of protein phosphatase 1 to F-actin in intact cells requires multiple domains in spinophilin and neurabin. FASEB J. 2008, 22, 1660–1671.
- Vivo, M.; Calogero, R.A.; Sansone, F.; Calabrò, V.; Parisi, T.; Borrelli, L.; Saviozzi, S.; La Mantia, G. The Human Tumor Suppressor ARF Interacts with Spinophilin/Neurabin II, a Type 1 Protein-phosphatase-binding Protein. J. Biol. Chem. 2001, 276, 14161–14169.
- Hansen, K.; Farkas, T.; Lukas, J.; Holm, K.; Rönnstrand, L.; Bartek, J. Phosphorylation-dependent and -independent functions of p130 cooperate to evoke a sustained G1 block. EMBO J. 2001, 20, 422–432.
- Tedesco, D.; Lukas, J.; Reed, S.I. The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2). Genes Dev. 2002, 16, 2946–2957.
- Fisher, L.A.; Wang, L.; Wu, L.; Peng, A. Phosphatase 1 nuclear targeting subunit is an essential regulator of M-phase entry, maintenance, and exit. J. Biol. Chem. 2014, 289, 23745–23752.
- Grossman, S.D.; Futter, M.; Snyder, G.L.; Allen, P.B.; Nairn, A.C.; Greengard, P.; Hsieh-Wilson, L.C. Spinophilin is phosphorylated by Ca2+/calmodulin-dependent protein kinase II resulting in regulation of its binding to F-actin. J. Neurochem. 2004, 90, 317–324.
- Futter, M.; Uematsu, K.; Bullock, S.A.; Kim, Y.; Hemmings, H.C.; Nishi, A.; Greengard, P.; Nairn, A.C. Phosphorylation of spinophilin by ERK and cyclin-dependent PK 5 (Cdk5). Proc. Natl. Acad. Sci. USA 2005, 102, 3489–3494.
- Hsieh-Wilson, L.C.; Benfenati, F.; Snyder, G.L.; Allen, P.B.; Nairn, A.C.; Greengard, P. Phosphorylation of spinophilin modulates its interaction with actin filaments. J. Biol. Chem. 2003, 278, 1186–1194.
- Edler, M.C.; Salek, A.B.; Watkins, D.S.; Kaur, H.; Morris, C.W.; Yamamoto, B.K.; Baucum, A.J. Mechanisms Regulating the Association of Protein Phosphatase 1 with Spinophilin and Neurabin. ACS Chem. Neurosci. 2018, 9, 2701–2712.
- Tsukada, M.; Prokscha, A.; Eichele, G. Neurabin II mediates doublecortin-dephosphorylation on actin filaments. Biochem. Biophys. Res. Commun. 2006, 343, 839–847.
- Shmueli, A.; Gdalyahu, A.; Sapoznik, S.; Sapir, T.; Tsukada, M.; Reiner, O. Site-specific dephosphorylation of doublecortin (DCX) by protein phosphatase 1 (PP1). Mol. Cell Neurosci. 2006, 32, 15–26.
- Bielas, S.L.; Serneo, F.F.; Chechlacz, M.; Deerinck, T.J.; Perkins, G.A.; Allen, P.B.; Ellisman, M.H.; Gleeson, J.G. Spinophilin Facilitates PP1-Mediated Dephosphorylation of PSer297 Doublecortin in Microtubule Bundling at the Axonal Wrist. Cell 2007, 129, 579–591.
- Abujiang, P.; Mori, T.J.; Takahashi, T.; Tanaka, F.; Kasyu, I.; Hitomi, S.; Hiai, H. Loss of heterozygosity (LOH) at 17q and 14q in human lung cancers. Oncogene 1998, 17, 3029–3033.
- Smith, S.A.; Easton, D.F.; Ford, D.; Peto, J.; Anderson, K.; Averill, D.; Stratton, M.; Ponder, M.; Pye, C.; Ponder, B.A. Genetic heterogeneity and localization of a familial breast-ovarian cancer gene on chromosome 17q12-q21. Am. J Hum. Genet. 1993, 52, 767–776.
- Caduff, R.F.; Svoboda-Newman, S.M.; Ferguson, A.W.; Frank, T.S. Comparison of alterations of chromosome 17 in carcinoma of the ovary and of the breast. Virchows Arch. 1999, 434, 517–522.
- Porter, D.E.; Steel, C.M.; Cohen, B.B.; Wallace, M.R.; Carothers, A.; Chetty, U.; Carter, D.C. Genetic linkage analysis applied to unaffected women from families with breast cancer can discriminate high- from low-risk individuals. Br. J. Surg. 1993, 80, 1381–1385.
- Porter, D.E.; Cohen, B.B.; Wallace, M.R.; Smyth, E.; Chetty, U.; Dixon, J.M.; Steel, C.M.; Carter, D.C. Breast cancer incidence, penetrance and survival in probable carriers of BRCA1 gene mutation in families linked to BRCA1 on chromosome 17q12-21. Br. J. Surg. 1994, 81, 1512–1515.
- Cohen, B.B.; Porter, D.E.; Wallace, M.R.; Carothers, A.; Steel, C.M. Linkage of a major breast cancer gene to chromosome 17q12-21: Results from 15 Edinburgh families. Am. J. Hum. Genet. 1993, 52, 723–729.
- Easton, D.F.; Bishop, D.T.; Ford, D.; Crockford, G.P. Genetic linkage analysis in familial breast and ovarian cancer: Results from 214 families. The Breast Cancer Linkage Consortium. Am. J. Hum Genet. 1993, 52, 678–701.
- Aigelsreiter, A.; Ress, A.L.; Bettermann, K.; Schauer, S.; Koller, K.; Eisner, F.; Kiesslich, T.; Stojakovic, T.; Samonigg, H.; Kornprat, P.; et al. Low expression of the putative tumour suppressor spinophilin is associated with higher proliferative activity and poor prognosis in patients with hepatocellular carcinoma. Br. J. Cancer 2013, 108, 1830–1837.
- Aigelsreiter, A.M.; Aigelsreiter, A.; Wehrschuetz, M.; Ress, A.L.; Koller, K.; Salzwimmer, M.; Gerger, A.; Schauer, S.; Bauernhofer, T.; Pichler, M. Loss of the putative tumor suppressor protein spinophilin is associated with poor prognosis in head and neck cancer. Hum. Pathol. 2014, 45, 683–690.
- Allen, P.B.; Zachariou, V.; Svenningsson, P.; Lepore, A.C.; Centonze, D.; Costa, C.; Rossi, S.; Bender, G.; Chen, G.; Feng, J.; et al. Distinct roles for spinophilin and neurabin in dopamine-mediated plasticity. Neuroscience 2006, 140, 897–911.
- Salek, A.B.; Edler, M.C.; McBride, J.P.; Baucum, A.J. Spinophilin regulates phosphorylation and interactions of the GluN2B subunit of the N-methyl-d-aspartate receptor. J. Neurochem. 2019, 151, 185–203.
- Feng, J.; Yan, Z.; Ferreira, A.; Tomizawa, K.; Liauw, J.A.; Zhuo, M.; Allen, P.B.; Ouimet, C.C.; Greengard, P. Spinophilin regulates the formation and function of dendritic spines. Proc. Natl. Acad. Sci. USA 2000, 97, 9287–9292.
- Jiang, Z.; Deng, T.; Jones, R.; Li, H.; Herschkowitz, J.I.; Liu, J.C.; Weigman, V.J.; Tsao, M.-S.; Lane, T.F.; Perou, C.M.; et al. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J. Clin. Investig. 2010, 120, 3296–3309.
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988.
- Carnero, A.; Garcia-Mayea, Y.; Mir, C.; Lorente, J.; Rubio, I.T.; LLeonart, M.E. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat Rev. 2016, 49, 25–36.
- Kareta, M.S.; Gorges, L.L.; Hafeez, S.; Benayoun, B.A.; Marro, S.; Zmoos, A.F.; Cecchini, M.J.; Spacek, D.; Batista, L.F.; O’Brien, M.; et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 2015, 16, 39–50.
- She, S.; Wei, Q.; Kang, B.; Wang, Y.-J. Cell cycle and pluripotency: Convergence on octamer-binding transcription factor 4. Mol. Med. Rep. 2017, 16, 6459–6466.
- Wang, M.-L.; Chiou, S.-H.; Wu, C.-W. Targeting cancer stem cells: Emerging role of Nanog transcription factor. OncoTargets Ther. 2013, 6, 1207–1220.
- Zhang, W.; Sui, Y.; Ni, J.; Yang, T. Insights into the Nanog gene: A propeller for stemness in primitive stem cells. Int. J. Biol. Sci. 2016, 12, 1372–1381.
- Schoeftner, S.; Scarola, M.; Comisso, E.; Schneider, C.; Benetti, R. An Oct4-pRb Axis, Controlled by MiR-335, Integrates Stem Cell Self-Renewal and Cell Cycle Control. Stem Cells 2013, 31, 717–728.
- Comisso, E.; Scarola, M.; Rosso, M.; Piazza, S.; Marzinotto, S.; Ciani, Y.; Orsaria, M.; Mariuzzi, L.; Schneider, C.; Schoeftner, S.; et al. OCT4 controls mitotic stability and inactivates the RB tumor suppressor pathway to enhance ovarian cancer aggressiveness. Oncogene 2017, 36, 4253–4266.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
792
Revisions:
2 times
(View History)
Update Date:
23 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No