 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giusy Di Conza | + 3363 word(s) | 3363 | 2021-05-19 11:35:09 | | | |
| 2 | Vivi Li | Meta information modification | 3363 | 2021-05-26 10:11:50 | | |
Video Upload Options
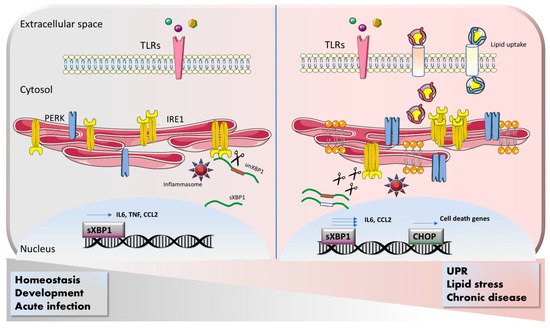
The endoplasmic reticulum (ER) is a critical organelle, storing the majority of calcium and governing protein translation. Thus, it is crucial to keep the homeostasis in all ER components and machineries. The ER stress sensor pathways, including IRE1/sXBP1, PERK/EIf2α and ATF6, orchestrate the major regulatory circuits to ensure ER homeostasis. The embryonic or postnatal lethality that occurs upon genetic depletion of these sensors reveals the essential role of the ER stress pathway in cell biology. In contrast, the impairment or excessive activation of ER stress has been reported to cause or aggravate several diseases such as atherosclerosis, diabetes, NAFDL/NASH, obesity and cancer. Being part of innate immunity, myeloid cells are the first immune cells entering the inflammation site. Upon entry into a metabolically stressed disease environment, activation of ER stress occurs within the myeloid compartment, leading to the modulation of their phenotype and functions.
1. Introduction
1.1. The Endoplasmic Reticulum and its Stress Sensors
1.2. ER Stress and Immune Signaling
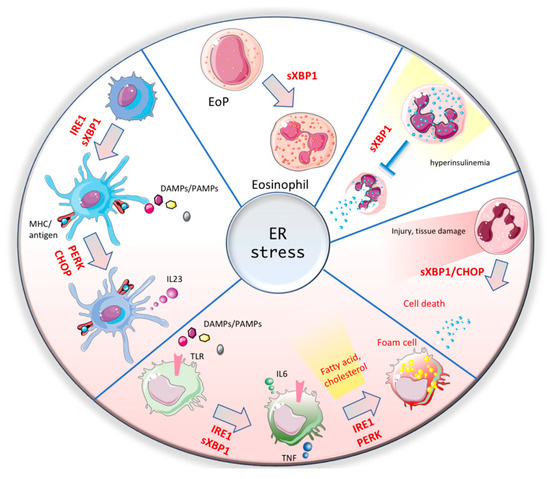
2. ER Stress Response in Myeloid Cells
2.1. Macrophages
2.2. Dendritic Cells
References
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94.
- Westrate, L.M.; Lee, J.E.; Prinz, W.A.; Voeltz, G.K. Form follows function: The importance of endoplasmic reticulum shape. Annu. Rev. Biochem. 2015, 84, 791–811.
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086.
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181.
- Volmer, R.; van der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633.
- Zhang, P.; McGrath, B.; Li, S.; Frank, A.; Zambito, F.; Reinert, J.; Gannon, M.; Ma, K.; McNaughton, K.; Cavener, D.R. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol. 2002, 22, 3864–3874.
- Zhang, W.; Feng, D.; Li, Y.; Iida, K.; McGrath, B.; Cavener, D.R. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006, 4, 491–497.
- Sovolyova, N.; Healy, S.; Samali, A.; Logue, S.E. Stressed to death—Mechanisms of ER stress-induced cell death. Biol. Chem. 2014, 395, 1–13.
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470.
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206.
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555.
- Glimcher, L.H. XBP1: The last two decades. Ann. Rheum. Dis. 2010, 69, 67–71.
- Jacquemyn, J.; Cascalho, A.; Goodchild, R.E. The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep. 2017, 18, 1905–1921.
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000, 14, 152–157.
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666.
- Lee, A.H.; Chu, G.C.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005, 24, 4368–4380.
- Iwawaki, T.; Akai, R.; Yamanaka, S.; Kohno, K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. USA 2009, 106, 16657–16662.
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376.
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; Garcia-Poyatos, C.; Martin-Garcia, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2alpha Axis. Mol. Cell 2019, 74, 877–890.
- Song, M.; Sandoval, T.A.; Chae, C.S.; Chopra, S.; Tan, C.; Rutkowski, M.R.; Raundhal, M.; Chaurio, R.A.; Payne, K.K.; Konrad, C.; et al. IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 2018, 562, 423–428.
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194.
- Binet, F.; Sapieha, P. ER Stress and Angiogenesis. Cell Metab. 2015, 22, 560–575.
- Li, S.; Wang, L.; Dorf, M.E. PKC phosphorylation of TRAF2 mediates IKKalpha/beta recruitment and K63-linked polyubiquitination. Mol. Cell 2009, 33, 30–42.
- Sakurai, H.; Suzuki, S.; Kawasaki, N.; Nakano, H.; Okazaki, T.; Chino, A.; Doi, T.; Saiki, I. Tumor necrosis factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J. Biol. Chem. 2003, 278, 36916–36923.
- Nishitoh, H.; Saitoh, M.; Mochida, Y.; Takeda, K.; Nakano, H.; Rothe, M.; Miyazono, K.; Ichijo, H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 1998, 2, 389–395.
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol. Cell. Biol. 2003, 23, 5651–5663.
- Bubici, C.; Papa, S.; Pham, C.G.; Zazzeroni, F.; Franzoso, G. NF-kappaB and JNK: An intricate affair. Cell Cycle 2004, 3, 1524–1529.
- Chen, C.; Zhang, X. IRE1alpha-XBP1 pathway promotes melanoma progression by regulating IL-6/STAT3 signaling. J. Transl. Med. 2017.
- Liu, Y.; Shao, M.; Wu, Y.; Yan, C.; Jiang, S.; Liu, J.; Dai, J.; Yang, L.; Li, J.; Jia, W.; et al. Role for the endoplasmic reticulum stress sensor IRE1alpha in liver regenerative responses. J. Hepatol. 2015, 62, 590–598.
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol. Cell. Biol. 2014, 34, 3911–3925.
- Chopra, S.; Giovanelli, P.; Alvarado-Vazquez, P.A.; Alonso, S.; Song, M.; Sandoval, T.A.; Chae, C.S.; Tan, C.; Fonseca, M.M.; Gutierrez, S.; et al. IRE1alpha-XBP1 signaling in leukocytes controls prostaglandin biosynthesis and pain. Science 2019.
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859.
- Geering, B.; Stoeckle, C.; Conus, S.; Simon, H.U. Living and dying for inflammation: Neutrophils, eosinophils, basophils. Trends Immunol. 2013, 34, 398–409.
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661.
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216.
- Lehner, P.J.; Cresswell, P. Recent developments in MHC-class-I-mediated antigen presentation. Curr. Opin. Immunol. 2004, 16, 82–89.
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chavez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R.; et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397.
- Martinon, F.; Chen, X.; Lee, A.H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418.
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nunez, G.; He, Y.; Yin, X.M.; O’Riordan, M.X. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity 2015, 43, 451–462.
- Qiu, Q.; Zheng, Z.; Chang, L.; Zhao, Y.S.; Tan, C.; Dandekar, A.; Zhang, Z.; Lin, Z.; Gui, M.; Li, X.; et al. Toll-like receptor-mediated IRE1alpha activation as a therapeutic target for inflammatory arthritis. EMBO J. 2013, 32, 2477–2490.
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850.
- Ray, I.; Mahata, S.K.; De, R.K. Obesity: An Immunometabolic Perspective. Front. Endocrinol 2016.
- Shan, B.; Wang, X.; Wu, Y.; Xu, C.; Xia, Z.; Dai, J.; Shao, M.; Zhao, F.; He, S.; Yang, L.; et al. The metabolic ER stress sensor IRE1alpha suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat. Immunol. 2017, 18, 519–529.
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343.
- Vega, H.; Agellon, L.B.; Michalak, M. The rise of proteostasis promoters. IUBMB Life 2016, 68, 943–954.
- Erbay, E.; Babaev, V.R.; Mayers, J.R.; Makowski, L.; Charles, K.N.; Snitow, M.E.; Fazio, S.; Wiest, M.M.; Watkins, S.M.; Linton, M.F.; et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat. Med. 2009, 15, 1383–1391.
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Cimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, 1395–1404.
- Petkevicius, K.; Virtue, S.; Bidault, G.; Jenkins, B.; Cubuk, C.; Morgantini, C.; Aouadi, M.; Dopazo, J.; Serlie, M.J.; Koulman, A.; et al. Accelerated phosphatidylcholine turnover in macrophages promotes adipose tissue inflammation in obesity. Elife 2019.
- Yan, D.; Wang, H.W.; Bowman, R.L.; Joyce, J.A. STAT3 and STAT6 Signaling Pathways Synergize to Promote Cathepsin Secretion from Macrophages via IRE1alpha Activation. Cell Rep. 2016, 16, 2914–2927.
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8.
- Thevenot, P.T.; Sierra, A.; Raber, P.L.; Al-Khami, A.A.; Trillo-Tinoco, J.; Zarreii, P.; Ochoa, A.C.; Cui, Y.; Del Valle, L.; Rodriguez, P.C. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity 2014, 41, 389–401.
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604.
- Blander, J.M. Regulation of the Cell Biology of Antigen Cross-Presentation. Annu. Rev. Immunol. 2018, 36, 717–753.
- Osorio, F.; Lambrecht, B.N.; Janssens, S. Antigen presentation unfolded: Identifying convergence points between the UPR and antigen presentation pathways. Curr. Opin. Immunol. 2018, 52, 100–107.
- Iwakoshi, N.N.; Pypaert, M.; Glimcher, L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007, 204, 2267–2275.
- Osorio, F.; Tavernier, S.J.; Hoffmann, E.; Saeys, Y.; Martens, L.; Vetters, J.; Delrue, I.; De Rycke, R.; Parthoens, E.; Pouliot, P.; et al. The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha+ dendritic cells. Nat. Immunol. 2014, 15, 248–257.
- Tavernier, S.J.; Osorio, F.; Vandersarren, L.; Vetters, J.; Vanlangenakker, N.; Van Isterdael, G.; Vergote, K.; De Rycke, R.; Parthoens, E.; van de Laar, L.; et al. Regulated IRE1-dependent mRNA decay sets the threshold for dendritic cell survival. Nat. Cell. Biol. 2017, 19, 698–710.
- Goodall, J.C.; Wu, C.; Zhang, Y.; McNeill, L.; Ellis, L.; Saudek, V.; Gaston, J.S. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17698–17703.
- Mogilenko, D.A.; Haas, J.T.; L’Homme, L.; Fleury, S.; Quemener, S.; Levavasseur, M.; Becquart, C.; Wartelle, J.; Bogomolova, A.; Pineau, L.; et al. Metabolic and Innate Immune Cues Merge into a Specific Inflammatory Response via the UPR. Cell 2019, 177, 1201–1216.
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538.
- Betts, B.C.; Locke, F.L.; Sagatys, E.M.; Pidala, J.; Walton, K.; Menges, M.; Reff, J.; Saha, A.; Djeu, J.Y.; Kiluk, J.V.; et al. Inhibition of Human Dendritic Cell ER Stress Response Reduces T Cell Alloreactivity Yet Spares Donor Anti-tumor Immunity. Front. Immunol. 2018.


