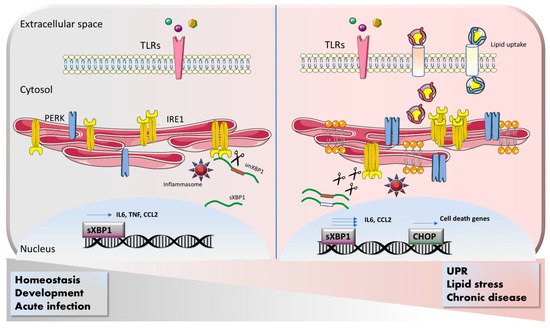
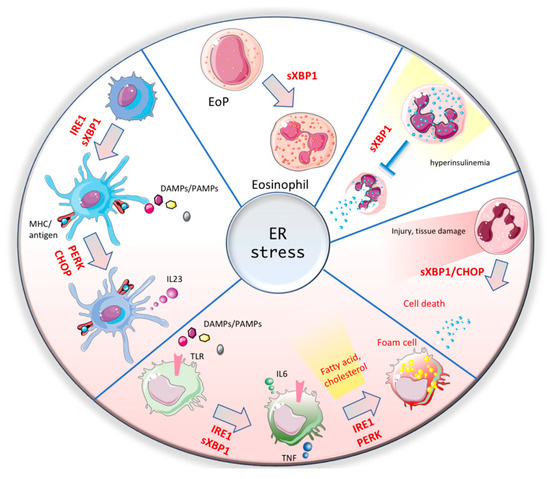
Dendritic cells (DCs) represent the connection arm between innate and adaptive immunity. DCs are able to cross-present antigens to CD8
+ and CD4
+ T cells, unleashing their activation and the engagement of antigen-specific immune responses. Based on location and functions, many subsets of DCs exist, varying from professional antigen-presenting cells to inflammatory DCs. Features and differences of DCs subsets have been nicely reviewed elsewhere [
52]. As other innate cells, DCs have myeloid origin and are activated by DAMPs and PAMPs through the expression of PRRs receptors. Once activated, antigen-loaded DCs migrate to draining lymph nodes where they encounter naïve T cells. Binding of the MHC/antigen complex to the T cell receptor in T cells leads to their activation and migration to the site of infection. The molecular mechanism orchestrating the process of antigen presentation is complex and to some extent still not completely uncovered [
53]. The ER host the loading of antigens on MHCI or MHCII complexes and for this reason many reports have demonstrated that the ER guardians are indirectly involved in the antigen processing and presentation [
54]. In one of the first papers showing the connection between ER stress and DCs, it was drastically proven that XBP1 deletion in the hematopoietic compartment impairs development and survival of dendritic cell lineage [
55]. Immature DCs constitutively activate XBP1, which in turn regulates their differentiation in a cell intrinsic manner. However, the molecular mechanism behind this crucial role of XBP1 was not elucidated. Later, more studies clarified that the severity of the phenotype upon genetic loss of XBP1 is different in distinct subsets. Splenic dendritic cells show impaired antigen presentation associated with a disturbed ER architecture. IRE1/XBP1 signaling is constitutively activated in splenic dendritic cells and promotes cross-presentation by directly regulating the expression of proteins crucial for the antigen loading into MHC complex [
56]. In contrast, conditional knockout of XBP1 under the CD11c promoter results in ablation of lung DCs via CHOP-mediated apoptosis. In the same mouse model, mucosal DCs survived. However, upon pharmacological blockade of IRE1 activity (which is overactivated in XBP1 KO), mucosal DCs also undergo cell death, suggesting a different threshold of IRE1 activation in different tissues [
57]. These studies have been performed in absence of infection and therefore are limited to the physiological status of DCs. However, as it occurs for macrophages, upon TLR activation, ER stress pathways are engaged in DCs and contribute to their function. Strikingly, treatment of DCs with TLR4 and TLR8 agonists induces CHOP activation and its direct binding to the promoter of the pro-inflammatory cytokine IL-23 [
58] (). Similarly, it has been recently published that palmitic acid or HFD are able to reinforce the effect of TLR7/8 agonist IMQ by boosting IL-23 production in DCs, upon activation of CHOP and sXBP1-mediated ER stress response [
59]. In cancer, dendritic cells are loaded with lipid that undergo peroxidation and induce an XBP1-mediated ER stress response. Conditional knockout mice harboring XBP1 deletion in dendritic cells were strongly protected against tumor growth compared to WT mice. Mechanistically, XBP1 boosts triglyceride production and further widens the overloading of lipids, therefore disturbing the machinery of antigen presentation and blunting anti-tumor response [
60]. In contrast, a study performed in human graft vs. host disease (GVHD), has shown that DCs-specific sXBP1 deletion strongly suppress alloreactive CD4 T cells, and pharmacological block of spliced XBP1 protects against rejection. Moreover, the authors provided evidence that this phenomenon is restrict to GVHD, because intratumoral T cell response was not affected by XBP1-expressing DCs [
61]. In conclusion, by directly or indirectly controlling antigen presentation in combination with TLR signaling, ER stress acts as major hub in regulating DCs functions and therefore represent an attracting therapeutic target. However, more studies are needed in order to fully understand how duration and intensity of inflammation and priming are interplaying with the ER guardians to balance physiologic versus pathologic DC responses.