 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gert Folkerts | + 2178 word(s) | 2178 | 2021-05-13 10:56:42 | | | |
| 2 | Camila Xu | -4 word(s) | 2174 | 2021-05-26 03:08:13 | | |
Video Upload Options
Chronic obstructive pulmonary disease (COPD) is a lung disease primarily characterized by the presence of airflow limitation and inflammation, due to elevated inflammatory cells, especially neutrophils, in the lungs.
1. Introduction
Chronic obstructive pulmonary disease (COPD) is a lung disease primarily characterized by the presence of airflow limitation and inflammation, due to elevated inflammatory cells, especially neutrophils, in the lungs [1]. Cigarette smoke (CS) is one of the major causes of COPD, which is responsible for chronic inflammation and mitochondria dysfunction in the lungs [2][3]. It is well known that mitochondria are an important source of reactive oxygen species (ROS) [4]. Decreased adenosine triphosphate (ATP) and increased ROS production in dysfunctional mitochondria cause an imbalance in intracellular homeostasis [5]. Recent studies identify changes in lung cell mitochondria, including a reduction in mitochondrial biogenesis, changes in mitochondrial DNA (mtDNA), selective degradation of mitochondria, and substantial morphological defects, as contributors to COPD pathogenesis [6][7][8][9][10][11].
Glucocorticoids, β2-adrenoceptor agonists, and muscarinic receptor antagonists are the major current therapy for COPD, which can reduce symptoms and/or exacerbations, but do not specifically target oxidative stress, nor do they diminish chronic respiratory inflammation and COPD progression or mortality in all patients [12][13]. A large case–control study indicated that treatment of COPD with a long-acting β2-agonist or a long-acting muscarinic antagonist was associated with a 50% higher risk of serious cardiovascular complications, including patients that had no cardiovascular disease history [14]. In addition, a large group of COPD patients have a poor response to glucocorticoids or are completely resistant [15]. Oxidative stress causes a reduction in histone deacetylase-2 (HDAC-2) activity and has been implicated as an important cause of steroid resistance in COPD [16][17]. Therefore, there is an urgent need for the development of novel therapies that effectively suppresses chronic inflammation in COPD patients.
Recently, a novel class of pharmacological compounds, 6-hydroxychromanols, were developed, which are also called SUL compounds. SULs are water-soluble Trolox-derivatives and accumulate in the mitochondria, displaying antioxidant and mitoprotective properties by alleviating ROS production and preserving ATP production [18]. Interestingly, SUL-121 (6-hydroxy-2,5,7,8-tetramethylchroman-2-yl (piperazin-1-yl) methanone), a racemic mix of SUL compounds dose-dependently reduced LPS-induced airway neutrophilia and airway hyperresponsiveness in guinea pigs. Furthermore, it also inhibited the cigarette smoke-induced interleukin 8 (IL-8) release accompanied by a decreased cellular ROS production in human airway smooth muscle cells [19].
In this entry, the efficacy of SUL-151 (Figure 1), the s-enantiomer of SUL-121, was examined in a prophylactic and therapeutic setting in mice triggered by CS to provoke oxidative stress and neutrophilic inflammation. Budesonide was used as a standard therapy to compare with the effectiveness of SUL-151 under the same conditions. Moreover, the mode of action of SUL-151 was explored. SUL-151 suppressed the CS-induced lung inflammation and mitochondrial dysfunction in this study, which appears as a promising candidate for the treatment of COPD to be assessed in future (pre) clinical trials.
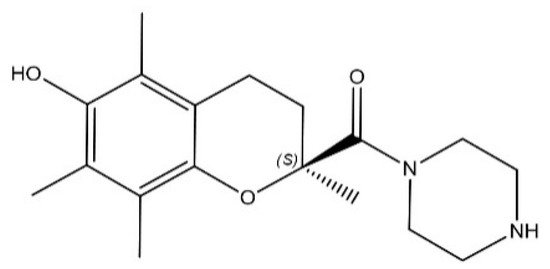
Figure 1. Chemical structure of SUL-151.
2. Prophylactic Treatment of SUL-151 Prevents Pulmonary Inflammation in a CS Exposure Model
SUL-151 concentrations in the lungs after 5 days of oropharyngeal administration to the lungs were on average 768.3 ± 101.2 pg/mg (ranged between 125 and 2264 pg/mg protein). The levels of SUL-151 in the serum were below the lower limit of detection (i.e., <5 pg/mL, data not shown).
CS exposure for 5 consecutive days significantly increased the number of total BALF cells (Figure 2A), macrophages (Figure 2B), neutrophils (Figure 2C), and BALF TNF-α level (Figure 2E), when compared to the air control group. SUL-151 administration for 5 days to the air-exposed animals did not significantly affect the number of neutrophils, lymphocytes, or TNF-α levels in BALF; however, it doubled the number of total cells in BALF, represented by an increase in macrophages.
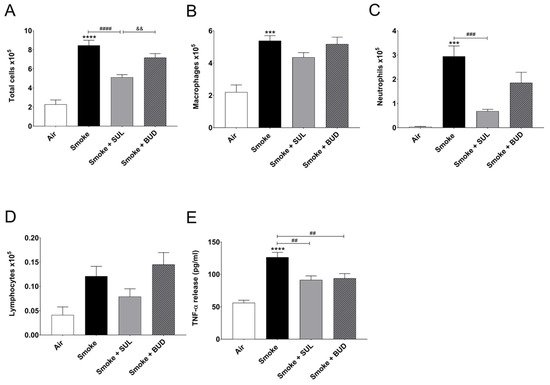
Figure 2. Prophylactic SUL-151 administration prevents pulmonary inflammation in a CS exposure model. Mice were exposed to CS for 5 days (twice/day) and received SUL-151 or budesonide via oropharyngeal administration (once/day) 30 min before the first-time smoke exposure during these 5 days. On day 6, lungs were lavaged, and BALF was collected for total (A) and differential BALF cell counts, including macrophages (B), neutrophils (C), and lymphocytes (D), and TNF-α levels (E) were measured in BALF. Values are expressed as mean ± SEM. *** p < 0.001, **** p < 0.0001, smoke group compared to air group; ## p < 0.01, ### p < 0.001, #### p < 0.0001, smoke + SUL group or smoke + budesonide (BUD) group compared to smoke group; && p < 0.01, smoke + BUD group compared to smoke + SUL group. n = 4–8 mice/group.
SUL-151 prophylaxis decreased the CS-induced increase in total cells in BALF (Figure 2A), wherein specifically neutrophilic infiltration was blunted (Figure 2C). Budesonide did not significantly affect the CS-induced increase of BALF total cells and neutrophils (Figure 2A,C). SUL-151 and budesonide did not affect the CS-induced increase in the numbers of macrophages and lymphocytes in BALF (Figure 2B,D); however, both compounds did reduce the TNF-α levels in BALF (Figure 2E).
3. Prophylactic Treatment of SUL-151 Prevents Oxidative Stress in the Lungs of CS-Exposed Mice
CS exposure for 5 consecutive days decreased the radical scavenging activity (Figure 3A) and increased the MDA levels (Figure 3B). In addition, a decrease of mtDNA (Figure 3C) and ATP (Figure 3D) was observed in the lung homogenates after CS exposure when compared to air-exposed mice. SUL-151 tended to increase the CS-induced decrease in radical scavenging capacity (p = 0.0771), but budesonide did not affect the CS-induced decrease in radical scavenging capacity (p = 0.2819) (Figure 3A), while both reduced the CS-induced increase in MDA in lung tissue (Figure 3B). SUL-151, but not budesonide, attenuated the loss of mitochondria by restoring the copy numbers of mtDNA (Figure 3C) and restored ATP production in lung tissue of CS-exposed mice (Figure 3D).
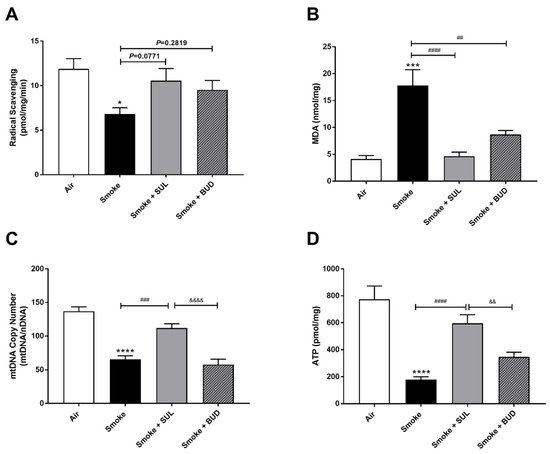
Figure 3. Prophylactic SUL-151 administration prevents oxidative stress in the lungs of a CS exposure model. Mice were exposed to CS for 5 days (twice/day) and received SUL-151 or budesonide via oropharyngeal administration 30 min before the first-time smoke exposure during these 5 days. On day 6, lung tissue was collected, and the radical scavenging activity (A), MDA concentration (B), mtDNA copy numbers (C), and ATP levels (D) were measured. Values are expressed as mean ± SEM. * p < 0.05, *** p < 0.001, **** p < 0.0001, smoke group compared to air group; ## p < 0.01, ### p < 0.001, #### p < 0.0001, smoke + SUL group or smoke + budesonide (BUD) group compared to smoke group; && p < 0.01, &&&& p < 0.0001, smoke + BUD group compared to the smoke + SUL group. n = 4–8 mice/group.
4. SUL-151 Reduces the Influx of Neutrophils in the BALF after the Development of CS-Induced Pulmonary Inflammation
Based on the promising effects of SUL-151 prophylaxis in the CS exposure model, the effect of SUL-151 was further explored in a therapeutic setting. Mice were first exposed to CS for 5 days without therapy, followed by 5 days of CS exposure with the same therapies as described for the prophylactic approach. The number of total BALF cells (Figure 4A), macrophages (Figure 4B), and neutrophils (Figure 4C) were significantly increased after 10 days of CS exposure, compared to the air-exposed group, while a slight effect of CS exposure was observed on the number of lymphocytes in BALF.
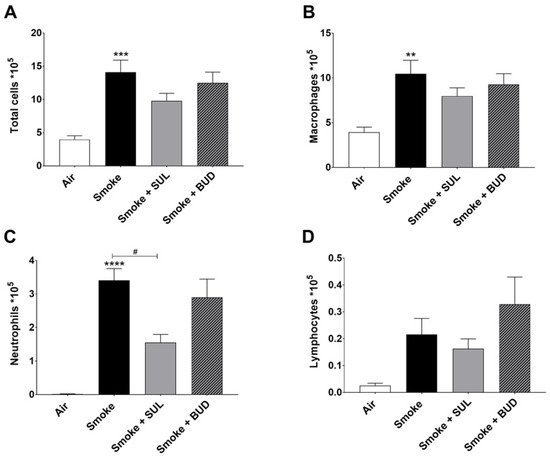
Figure 4. SUL-151 reduces the influx of neutrophils in the BALF after the development of CS-induced pulmonary inflammation. Mice were exposed to CS for 10 days (twice/day) and received SUL-151 or budesonide via oropharyngeal administration (once/day) 30 min before the first-time smoke exposure during the last 5 days of the CS exposure period. On day 11, lungs were lavaged, and BALF was collected for total (A) and differential BAL cell counts, including macrophages (B), neutrophils (C), and lymphocytes (D). Values are expressed as mean ± SEM. ** p < 0.01, *** p < 0.001, **** p < 0.0001, smoke group compared to air group; # p < 0.05, smoke + SUL group or smoke + budesonide (BUD) group compared to smoke group. n = 6–7 mice/group.
Both the treatment with SUL-151 and budesonide did not significantly affect the CS-induced increase in the number of total cells, macrophages, and lymphocytes in the BALF (Figure 4A,B,D). No differences were observed in TNF-α levels in BALF (data not shown). SUL-151, but not budesonide, was able to significantly reduce the CS-induced influx of neutrophils in the second set of 5 days CS exposure (day 10, Figure 4C) to a similar degree as in the prophylactic setting (Figure 2C).
5. SUL-151 Reduces the Oxidative Stress in the Lungs after the Development of CS-Induced Pulmonary Inflammation
The radical scavenging activity was decreased (Figure 5A), and MDA levels were increased (Figure 5C) in the lungs after CS exposure. Moreover, the mtDNA copy numbers (Figure 5E) and the ATP levels (Figure 5F) were decreased in the lung homogenates 10 days after CS exposure. As observed in the prophylactic setting, SUL-151 tended to restore the CS-induced changes in the radical scavenging ability (p = 0.0513, Figure 5A), and both SUL-151 and budesonide decreased MDA levels in the lungs obtained from the mice of the therapeutic approach (Figure 5C). The increase in scavenging activity and the reduction of lipid peroxidation products observed in individual mice correlated to the concentration of SUL-151 in the lung homogenates (Figure 5B,D, respectively). Interestingly, SUL-151, significantly restored mtDNA copy numbers and ATP levels in the CS-exposed mice, while budesonide was not effective at restoring mtDNA or ATP levels (Figure 5E,F).
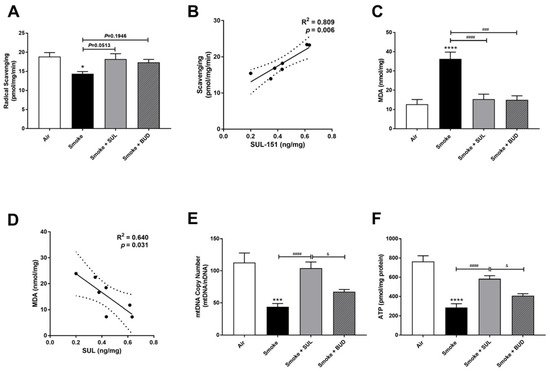
Figure 5. SUL-151 reduces the oxidative stress in the lungs after the development of CS-induced pulmonary inflammation. Mice were exposed to CS for 10 days (twice/day) and received SUL-151 or budesonide via oropharyngeal administration (once/day) 30 min before the first smoke exposure during the last 5 days of the CS exposure period. On day 11, lung tissue was collected, and the radical scavenging activity (A), MDA concentration (C), mtDNA copy numbers (E), and ATP levels (F) were measured. Correlation of radical scavenging activity and SUL-151 concentration in lung tissue (B) and the correlation of MDA and SUL-151 concentration in lung tissue (D) analyzed using Pearson r correlation test. Values are expressed as mean ± SEM. * p < 0.05, *** p < 0.001, **** p < 0.0001, smoke group compared to air group; ### p < 0.001, #### p < 0.0001, smoke + SUL group or smoke + budesonide (BUD) group compared to smoke group; & p < 0.05, smoke + BUD group compared to smoke + SUL group. n = 6–7 mice/group.
6. SUL-151 Inhibits the Increase in PINK1-Expression in the Lungs after the Development of CS-Induced Pulmonary Inflammation
Parkin, a ubiquitin–protein ligase, and PTEN-induced putative kinase 1 (PINK1), a mitochondrial serine–threonine kinase, both exhibit protection against oxidative stress and act in a common mitophagy pathway [20][21]. The expression of PINK1 was significantly increased after 10 days of CS exposure. SUL-151, but not budesonide, significantly inhibited the CS-induced increase in PINK1 protein expression (Figure 6A,B). CS did not significantly change the Parkin protein expression in lung tissue (Figure 6A,C), while budesonide, but not SUL-151, further decreased the Parkin protein expression compared with the CS-exposed group.
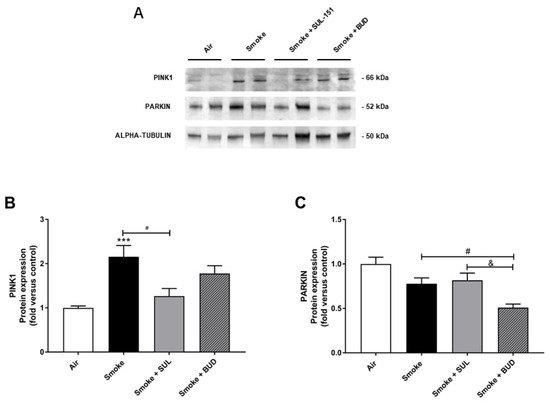
Figure 6. SUL-151 inhibited the increase in PINK1-expression in the lungs after the development of CS-induced pulmonary inflammation. Mice were exposed to CS for 10 days (twice/day) and received SUL-151 or budesonide via oropharyngeal administration (once/day) 30 min before the first smoke exposure during the last 5 days of the CS period. Western blot analysis for protein levels of PINK1, Parkin, and α-tubulin (A) was measured in the lung homogenates, and relative densities of the PINK1/α-tubulin (B) and Parkin /α-tubulin (C) were calculated. Values are expressed as mean ± SEM. *** p < 0.001, smoke group compared to air group; # p < 0.05, smoke + SUL group or smoke + budesonide (BUD) group compared to smoke group; & p < 0.05, smoke + BUD group compared to smoke +SUL group. n = 6–7 mice/group.
7. SUL-151 Hardly Affects KC Levels in Lung Homogenates but Concentration-Dependently Inhibits IL8-Production in Human Bronchial Epithelial Cells
To find a link between the reduced oxidative stress and decrease in BALF neutrophil numbers, KC levels were measured in lung homogenates. Indeed, CS induced an increase in KC levels in the lungs. However, this CS-induced increase in KC levels was hardly affected by SUL-151 or budesonide as a prophylactic or therapeutic treatment (Figure 7A,B).
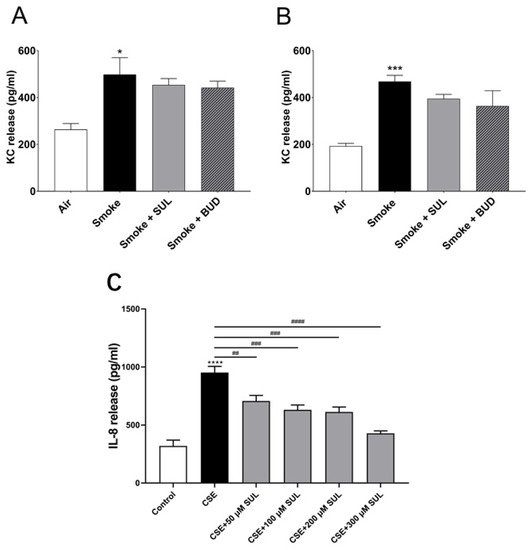
Figure 7. SUL-151 hardly affects KC levels in lung homogenates but concentration-dependently inhibits IL-8 production in human bronchial epithelial cells. Mice were exposed to CS for 5 days (twice/day) and received SUL-151 or budesonide via oropharyngeal administration 30 min before the first-time smoke exposure during these 5 days (A) or mice were exposed to CS for 10 days (twice/day) and received SUL-151 or budesonide via oropharyngeal administration (once/day) 30 min before the first smoke exposure during the last 5 days of the CS period (B). KC levels were measured in the lung homogenates via ELISA measurement. Human bronchial epithelial cells (16HBE cells), grown on 96-well plates, were preincubated with different concentrations of SUL-151 (50, 100, 200, and 300 µM) (24h) prior to CSE exposure for 24 h. Thereafter, IL-8 secretion was measured in the supernatants (C). Values are expressed as mean ± SEM. * p < 0.05, *** p < 0.001, **** p < 0.0001, smoke/CSE group compared to air/control group; ## p < 0.01, ### p < 0.001, #### p < 0.0001, CSE group compared to the CSE + SUL group. n = 4–8 mice/group in vivo, n = 5, in vitro.
To further explore the effect of SUL-151 on IL-8 production, human bronchial epithelial (16HBE) cells were stimulated with cigarette smoke extract (CSE). IL-8 production was significantly enhanced after CSE exposure for 24 h. Surprisingly, SUL-151 concentration-dependently decreased the CSE-induced IL-8 production (Figure 7C).
References
- Eapen, M.S.; Myers, S.; Walters, E.H.; Sohal, S.S. Airway inflammation in chronic obstructive pulmonary disease (COPD): A true paradox. Expert Rev. Respir. Med. 2017, 11, 827–839.
- Lundbäck, B.; Lindberg, A.; Lindström, M.; Rönmark, E.; Jonsson, A.; Jönsson, E.; Larsson, L.-G.; Andersson, S.; Sandström, T.; Larsson, K. Not 15 but 50% of smokers develop COPD?—Report from the Obstructive Lung Disease in Northern Sweden Studies. Respir. Med. 2003, 97, 115–122.
- Tsubouchi, K.; Araya, J.; Kuwano, K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm. Regen. 2018, 38, 1–9.
- Dunn, J.D.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485.
- Hara, H.; Kuwano, K.; Araya, J. Mitochondrial Quality Control in COPD and IPF. Cells 2018, 7, 86.
- Zhang, L.; Wang, W.; Zhu, B.; Wang, X. Epithelial Mitochondrial Dysfunction in Lung Disease. Chem. Biol. Pteridines Folates 2017, 1038, 201–217.
- Jiang, Y.; Wang, X.; Hu, D. Mitochondrial alterations during oxidative stress in chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 1153–1162.
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; De Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97.
- Yue, L.; Yao, H. Mitochondrial dysfunction in inflammatory responses and cellular senescence: Pathogenesis and pharmacological targets for chronic lung diseases. Br. J. Pharmacol. 2016, 173, 2305–2318.
- Liu, S.-F.; Kuo, H.-C.; Tseng, C.-W.; Huang, H.-T.; Chen, Y.-C.; Tseng, C.-C.; Lin, M.-C. Leukocyte Mitochondrial DNA Copy Number Is Associated with Chronic Obstructive Pulmonary Disease. PLoS ONE 2015, 10, e0138716.
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J. 2015, 29, 2912–2929.
- Ernst, P.; Saad, N.; Suissa, S. Inhaled corticosteroids in COPD: The clinical evidence. Eur. Respir. J. 2014, 45, 525–537.
- Barnes, P.J. COPD 2020: New directions needed. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L884–L886.
- Wang, M.T.; Liou, J.T.; Lin, C.W.; Tsai, C.L.; Wang, Y.H.; Hsu, Y.J.; Lai, J.H. Association of Cardiovascular Risk with Inhaled Long-Acting Bronchodilators in Patients with Chronic Obstructive Pulmonary Disease: A Nested Case-Control Study. JAMA Intern. Med. 2018, 178, 229–238.
- Jiang, Z.; Zhu, L. Update on molecular mechanisms of corticosteroid resistance in chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2016, 37, 1–8.
- Barnes, P.J.; Adcock, I.M. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917.
- Marwick, J.A.; Ito, K.; Adcock, I.M.; A Kirkham, P. Oxidative stress and steroid resistance in asthma and COPD: Pharmacological manipulation of HDAC-2 as a therapeutic strategy. Expert Opin. Ther. Targets 2007, 11, 745–755.
- Hajmousa, G.; Vogelaar, P.; Brouwer, L.A.; Van Der Graaf, A.C.; Henning, R.H.; Krenning, G. The 6-chromanol derivate SUL-109 enables prolonged hypothermic storage of adipose tissue-derived stem cells. Biomaterials 2017, 119, 43–52.
- Han, B.; Poppinga, W.J.; Zuo, H.; Zuidhof, A.B.; Bos, I.S.T.; Smit, M.; Vogelaar, P.; Krenning, G.; Henning, R.H.; Maarsingh, H.; et al. The novel compound Sul-121 inhibits airway inflammation and hyperresponsiveness in experimental models of chronic obstructive pulmonary disease. Sci. Rep. 2016, 6, 26928.
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643.
- Ashrafi, G.H.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42.


