 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jorge E. Contreras | + 3280 word(s) | 3280 | 2021-05-17 08:02:25 | | | |
| 2 | Vivi Li | Meta information modification | 3280 | 2021-05-25 08:06:21 | | |
Video Upload Options
Neuroinflammation is a major component of central nervous system (CNS) injuries and neurological diseases, including Alzheimer’s disease, multiple sclerosis, neuropathic pain, and brain trauma. The activation of innate immune cells at the damage site causes the release of pro-inflammatory cytokines and chemokines, which alter the functionality of nearby tissues and might mediate the recruitment of leukocytes to the injury site. If this process persists or is exacerbated, it prevents the adequate resolution of the inflammation, and ultimately enhances secondary damage. Adenosine 5′ triphosphate (ATP) is among the molecules released that trigger an inflammatory response, and it serves as a chemotactic and endogenous danger signal. Extracellular ATP activates multiple purinergic receptors (P2X and P2Y) that have been shown to promote neuroinflammation in a variety of CNS diseases. Recent studies have shown that Pannexin-1 (Panx1) channels are the principal conduits of ATP release from dying cells and innate immune cells in the brain.
1. Background
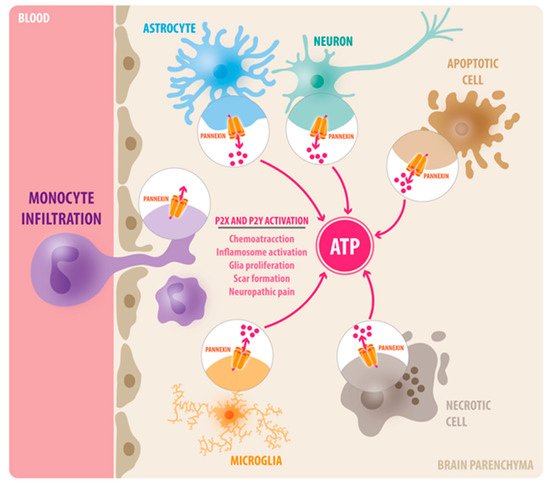
2. Pannexin-1 (Panx1) Channels
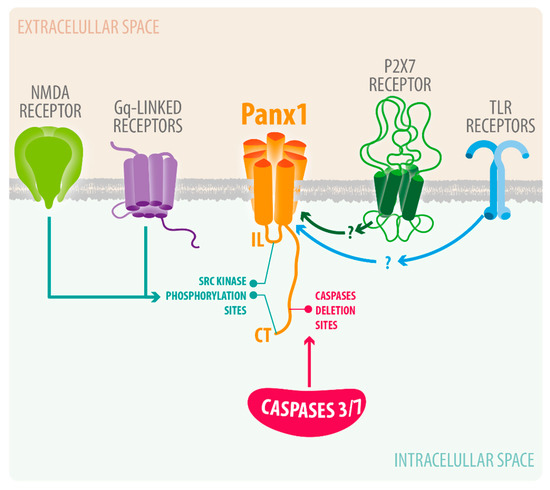
3. Pannexin-1 Channels in Neuroinflammation
| Pathophysiology | Pharmacological Blockade of Panx1 | Genetic Deletion | Outcome |
|---|---|---|---|
| Epilepsy | Probenecid and mefloquine | Global Panx1 KO | Blocked Ictal discharge and resistance to Kainic induced seizure (Dossi et al., 2018) |
| Spinal cord injury | 10Panx, Mefloquine, Probenecid | Microglia Panx1 KO | Reduces Morphine withdrawal and joint pain by mechanical allodynia (Mousseau et al., 2018) (Burma et al., 2017) |
| Sciatic Nerve injury (Neuropathic pain) | Carbenoxolone and Trovafloxacin | Global Panx1 KO Myeloid Panx1 (Microglia and infiltrating monocytes) KO |
Blockers reduced hypersensitivity to tactile and thermal stimuli Myeloid Panx1 KO did not reduced neuropathic pain (Weaver et al., 2017) |
| Ischemia (MCAO) | Probenecid | Global Panx1 KO Double KO of Panx1 and Panx2 |
Probenecid reduced neuronal death and inflammasome activation in rat model of ischemia.(Wei et al., 2015) Panx1 KO did not show neuroprotection but Double KO of Panx1 and Panx2 showed reduced neurological deficits and reduced infarct volume compared to WT in MCAO model (Bargiotas et al., 2011) |
| Ischemia/Reperfusion | Probenecid, mefloquine, Carbenoxolone | Global Panx1 KO | Blockers as well as Panx1 KO showed reduced infarct volume, neuronal and tissue damage and improved motor function. (Cisneros-Mejorado et al., 2015) |
| Traumatic brain injury | Trovafloxacin(TVX) | Myeloid Panx1 KO | TVX reduced macrophage infiltration and astrogliosis correlated with improvement in locomotor activity(Garg et al., 2018) Myeloid Panx1 KO showed improved motor co-ordination, memory outcomes, reduced tissue damage, less BBB leakage and less infiltration of leukocytes (Seo et al., 2020) |
| Experimental Autoimmune Encephalopathy | Mefloquine | Global Panx1 KO | Panx1 KO mice showed delayed onset of clinical signs of EAE and decreased mortality rate compared to WT mice Mefloquine (MFQ) reduced severity of acute and chronic EAE (Lutz, S.E., et al., 2013) |
3.1. Spinal Cord Injury, Neuropathic Pain, and Orofacial Pain
3.2. Brain Ischemia
3.3. Traumatic Brain Injury
References
- Cova, I.; Markova, A.; Campini, I.; Grande, G.; Mariani, C.; Pomati, S. Worldwide trends in the prevalence of dementia. J. Neurol. Sci. 2017, 379, 259–260.
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart disease and stroke statistics—2020 update: A report from the American Heart Association. Circulation 2020, 141, E139–E596.
- Taylor, C.A.; Bell, J.M.; Breiding, M.J.; Xu, L. Traumatic brain injury-related emergency department visits, hospitalizations, and deaths—United States, 2007 and 2013. MMWR Surveill. Summ. 2017, 66, 1–16.
- Kacirova, M.; Zmeskalova, A.; Korinkova, L.; Zelezna, B.; Kunes, J.; Maletinska, L. Inflammation: Major denominator of obesity, Type 2 diabetes and Alzheimer’s disease-like pathology? Clin. Sci. 2020, 134, 547–570.
- Hur, J.Y.; Frost, G.R.; Wu, X.; Crump, C.; Pan, S.J.; Wong, E.; Barros, M.; Li, T.; Nie, P.; Zhai, Y.; et al. The innate immunity protein IFITM3 modulates gamma-secretase in Alzheimer’s disease. Nature 2020, 586, 735–740.
- Iannucci, J.; Renehan, W.; Grammas, P. Thrombin, a Mediator of Coagulation, Inflammation, and Neurotoxicity at the Neurovascular Interface: Implications for Alzheimer’s Disease. Front. Neurosci. 2020, 14, 762.
- Ziebell, J.M.; Rowe, R.K.; Muccigrosso, M.M.; Reddaway, J.T.; Adelson, P.D.; Godbout, J.P.; Lifshitz, J. Aging with a traumatic brain injury: Could behavioral morbidities and endocrine symptoms be influenced by microglial priming? Brain Behav. Immun. 2017, 59, 1–7.
- Anrather, J.; Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 2016, 13, 661–670.
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489.
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633.
- Bruzzone, R.; Hormuzdi, S.G.; Barbe, M.T.; Herb, A.; Monyer, H. Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13644–13649.
- Dvoriantchikova, G.; Ivanov, D.; Panchin, Y.; Shestopalov, V.I. Expression of pannexin family of proteins in the retina. FEBS Lett. 2006, 580, 2178–2182.
- Dvoriantchikova, G.; Ivanov, D.; Pestova, A.; Shestopalov, V. Molecular characterization of pannexins in the lens. Mol. Vis. 2006, 12, 1417–1426.
- Ray, A.; Zoidl, G.; Wahle, P.; Dermietzel, R. Pannexin expression in the cerebellum. Cerebellum 2006, 5, 189–192.
- Ray, A.; Zoidl, G.; Weickert, S.; Wahle, P.; Dermietzel, R. Site-specific and developmental expression of pannexin1 in the mouse nervous system. Eur. J. Neurosci. 2005, 21, 3277–3290.
- Sohl, G.; Maxeiner, S.; Willecke, K. Expression and functions of neuronal gap junctions. Nat. Rev. Neurosci. 2005, 6, 191–200.
- Shestopalov, V.I.; Panchin, Y. Pannexins and gap junction protein diversity. Cell Mol. Life Sci. 2008, 65, 376–394.
- Vogt, A.; Hormuzdi, S.G.; Monyer, H. Pannexin1 and Pannexin2 expression in the developing and mature rat brain. Brain Res. Mol. Brain Res. 2005, 141, 113–120.
- Weickert, S.; Ray, A.; Zoidl, G.; Dermietzel, R. Expression of neural connexins and pannexin1 in the hippocampus and inferior olive: A quantitative approach. Brain Res. Mol. Brain Res. 2005, 133, 102–109.
- Zappala, A.; Cicero, D.; Serapide, M.F.; Paz, C.; Catania, M.V.; Falchi, M.; Parenti, R.; Panto, M.R.; La Delia, F.; Cicirata, F. Expression of pannexin1 in the CNS of adult mouse: Cellular localization and effect of 4-aminopyridine-induced seizures. Neuroscience 2006, 141, 167–178.
- Zoidl, G.; Petrasch-Parwez, E.; Ray, A.; Meier, C.; Bunse, S.; Habbes, H.W.; Dahl, G.; Dermietzel, R. Localization of the pannexin1 protein at postsynaptic sites in the cerebral cortex and hippocampus. Neuroscience 2007, 146, 9–16.
- Penuela, S.; Gehi, R.; Laird, D.W. The biochemistry and function of pannexin channels. Biochim. Biophys. Acta 2013, 1828, 15–22.
- Saez, P.J.; Vargas, P.; Shoji, K.F.; Harcha, P.A.; Lennon-Dumenil, A.M.; Saez, J.C. ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X7 receptors. Sci. Signal. 2017, 10, eaah7107.
- Seo, J.H.; Dalal, M.S.; Calderon, F.; Contreras, J.E. Myeloid Pannexin-1 mediates acute leukocyte infiltration and leads to worse outcomes after brain trauma. J. Neuroinflamm. 2020, 17, 245.
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31.
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867.
- Bravo, D.; Ibarra, P.; Retamal, J.; Pelissier, T.; Laurido, C.; Hernandez, A.; Constandil, L. Pannexin 1: A novel participant in neuropathic pain signaling in the rat spinal cord. Pain 2014, 155, 2108–2115.
- Zhang, Y.; Laumet, G.; Chen, S.R.; Hittelman, W.N.; Pan, H.L. Pannexin-1 Up-regulation in the Dorsal Root Ganglion Contributes to Neuropathic Pain Development. J. Biol. Chem. 2015, 290, 14647–14655.
- Chiu, Y.H.; Schappe, M.S.; Desai, B.N.; Bayliss, D.A. Revisiting multimodal activation and channel properties of Pannexin 1. J. Gen. Physiol. 2018, 150, 19–39.
- Weilinger, N.L.; Lohman, A.W.; Rakai, B.D.; Ma, E.M.; Bialecki, J.; Maslieieva, V.; Rilea, T.; Bandet, M.V.; Ikuta, N.T.; Scott, L.; et al. Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat. Neurosci. 2016, 19, 432–442.
- Lohman, A.W.; Leskov, I.L.; Butcher, J.T.; Johnstone, S.R.; Stokes, T.A.; Begandt, D.; DeLalio, L.J.; Best, A.K.; Penuela, S.; Leitinger, N.; et al. Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat. Commun. 2015, 6, 7965.
- Sanchez-Arias, J.C.; van der Slagt, E.; Vecchiarelli, H.A.; Candlish, R.C.; York, N.; Young, P.A.; Shevtsova, O.; Juma, A.; Tremblay, M.E.; Swayne, L.A. Purinergic signaling in nervous system health and disease: Focus on pannexin 1. Pharmacol. Ther. 2021, 107840.
- Ishikawa, M.; Williams, G.L.; Ikeuchi, T.; Sakai, K.; Fukumoto, S.; Yamada, Y. Pannexin 3 and connexin 43 modulate skeletal development through their distinct functions and expression patterns. J. Cell Sci. 2016, 129, 1018–1030.
- Zhang, P.; Ishikawa, M.; Doyle, A.; Nakamura, T.; He, B.; Yamada, Y. Pannexin 3 regulates skin development via Epiprofin. Sci. Rep. 2021, 11, 1779.
- Scemes, E.; Spray, D.C.; Meda, P. Connexins, pannexins, innexins: Novel roles of “hemi-channels”. Pflug. Arch. 2009, 457, 1207–1226.
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082.
- Illes, P. P2X7 Receptors Amplify CNS Damage in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5996.
- Freeman, T.J.; Sayedyahossein, S.; Johnston, D.; Sanchez-Pupo, R.E.; O’Donnell, B.; Huang, K.; Lakhani, Z.; Nouri-Nejad, D.; Barr, K.J.; Harland, L.; et al. Inhibition of Pannexin 1 Reduces the Tumorigenic Properties of Human Melanoma Cells. Cancers 2019, 11, 102.
- Graham, S.V.; Jiang, J.X.; Mesnil, M. Connexins and Pannexins: Important Players in Tumorigenesis, Metastasis and Potential Therapeutics. Int. J. Mol. Sci. 2018, 19, 1645.
- Thompson, R.J.; Zhou, N.; MacVicar, B.A. Ischemia opens neuronal gap junction hemichannels. Science 2006, 312, 924–927.
- Taylor, K.A.; Wright, J.R.; Vial, C.; Evans, R.J.; Mahaut-Smith, M.P. Amplification of human platelet activation by surface pannexin-1 channels. J. Thromb. Haemost. 2014, 12, 987–998.
- Dossi, E.; Blauwblomme, T.; Moulard, J.; Chever, O.; Vasile, F.; Guinard, E.; Le Bert, M.; Couillin, I.; Pallud, J.; Capelle, L.; et al. Pannexin-1 channels contribute to seizure generation in human epileptic brain tissue and in a mouse model of epilepsy. Sci. Transl. Med. 2018, 10, eaar3796.
- Orellana, J.A.; Velasquez, S.; Williams, D.W.; Saez, J.C.; Berman, J.W.; Eugenin, E.A. Pannexin1 hemichannels are critical for HIV infection of human primary CD4+ T lymphocytes. J. Leukoc. Biol. 2013, 94, 399–407.
- Scemes, E.; Velisek, L.; Veliskova, J. Astrocyte and Neuronal Pannexin1 Contribute Distinctly to Seizures. ASN Neuro 2019, 11.
- Santiago, M.F.; Veliskova, J.; Patel, N.K.; Lutz, S.E.; Caille, D.; Charollais, A.; Meda, P.; Scemes, E. Targeting pannexin1 improves seizure outcome. PLoS ONE 2011, 6, e25178.
- Allan, S.M.; Rothwell, N.J. Inflammation in central nervous system injury. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 1669–1677.
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487.
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142.
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554.
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716.
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938.
- Garre, J.M.; Retamal, M.A.; Cassina, P.; Barbeito, L.; Bukauskas, F.F.; Saez, J.C.; Bennett, M.V.; Abudara, V. FGF-1 induces ATP release from spinal astrocytes in culture and opens pannexin and connexin hemichannels. Proc. Natl. Acad. Sci. USA 2010, 107, 22659–22664.
- Burma, N.E.; Bonin, R.P.; Leduc-Pessah, H.; Baimel, C.; Cairncross, Z.F.; Mousseau, M.; Shankara, J.V.; Stemkowski, P.L.; Baimoukhametova, D.; Bains, J.S.; et al. Blocking microglial pannexin-1 channels alleviates morphine withdrawal in rodents. Nat. Med. 2017, 23, 355–360.
- Weaver, J.L.; Arandjelovic, S.; Brown, G.; Mendu, S.K.; Schappe, M.S.; Buckley, M.W.; Chiu, Y.H.; Shu, S.; Kim, J.K.; Chung, J.; et al. Hematopoietic pannexin 1 function is critical for neuropathic pain. Sci. Rep. 2017, 7, 42550.
- Garre, J.M.; Yang, G.; Bukauskas, F.F.; Bennett, M.V. FGF-1 Triggers Pannexin-1 Hemichannel Opening in Spinal Astrocytes of Rodents and Promotes Inflammatory Responses in Acute Spinal Cord Slices. J. Neurosci. 2016, 36, 4785–4801.
- Lutz, S.E.; Gonzalez-Fernandez, E.; Ventura, J.C.; Perez-Samartin, A.; Tarassishin, L.; Negoro, H.; Patel, N.K.; Suadicani, S.O.; Lee, S.C.; Matute, C.; et al. Contribution of pannexin1 to experimental autoimmune encephalomyelitis. PLoS ONE 2013, 8, e66657.
- Mousseau, M.; Burma, N.E.; Lee, K.Y.; Leduc-Pessah, H.; Kwok, C.H.T.; Reid, A.R.; O’Brien, M.; Sagalajev, B.; Stratton, J.A.; Patrick, N.; et al. Microglial pannexin-1 channel activation is a spinal determinant of joint pain. Sci. Adv. 2018, 4, eaas9846.
- Hanstein, R.; Negoro, H.; Patel, N.K.; Charollais, A.; Meda, P.; Spray, D.C.; Suadicani, S.O.; Scemes, E. Promises and pitfalls of a Pannexin1 transgenic mouse line. Front. Pharmacol. 2013, 4, 61.
- Chichorro, J.G.; Porreca, F.; Sessle, B. Mechanisms of craniofacial pain. Cephalalgia 2017, 37, 613–626.
- Karatas, H.; Erdener, S.E.; Gursoy-Ozdemir, Y.; Lule, S.; Eren-Kocak, E.; Sen, Z.D.; Dalkara, T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013, 339, 1092–1095.
- Hanstein, R.; Hanani, M.; Scemes, E.; Spray, D.C. Glial pannexin1 contributes to tactile hypersensitivity in a mouse model of orofacial pain. Sci. Rep. 2016, 6, 38266.
- Charles, A.C.; Baca, S.M. Cortical spreading depression and migraine. Nat. Rev. Neurol. 2013, 9, 637–644.
- Moskowitz, M.A. The neurobiology of vascular head pain. Ann. Neurol. 1984, 16, 157–168.
- Moskowitz, M.A.; Nozaki, K.; Kraig, R.P. Neocortical spreading depression provokes the expression of c-fos protein-like immunoreactivity within trigeminal nucleus caudalis via trigeminovascular mechanisms. J. Neurosci. 1993, 13, 1167–1177.
- Dalkara, T.; Zervas, N.T.; Moskowitz, M.A. From spreading depression to the trigeminovascular system. Neurol. Sci. 2006, 27 (Suppl. 2), S86–S90.
- Zhang, X.; Levy, D.; Noseda, R.; Kainz, V.; Jakubowski, M.; Burstein, R. Activation of meningeal nociceptors by cortical spreading depression: Implications for migraine with aura. J. Neurosci. 2010, 30, 8807–8814.
- Bolay, H.; Reuter, U.; Dunn, A.K.; Huang, Z.; Boas, D.A.; Moskowitz, M.A. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat. Med. 2002, 8, 136–142.
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385.
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Primers 2019, 5, 70.
- Jin, R.; Liu, L.; Zhang, S.; Nanda, A.; Li, G. Role of inflammation and its mediators in acute ischemic stroke. J. Cardiovasc. Transl. Res. 2013, 6, 834–851.
- Santos, M.S.; Moreno, A.J.; Carvalho, A.P. Relationships between ATP depletion, membrane potential, and the release of neurotransmitters in rat nerve terminals. An in vitro study under conditions that mimic anoxia, hypoglycemia, and ischemia. Stroke 1996, 27, 941–950.
- Weilinger, N.L.; Tang, P.L.; Thompson, R.J. Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J. Neurosci. 2012, 32, 12579–12588.
- Ogura, Y.; Sutterwala, F.S.; Flavell, R.A. The inflammasome: First line of the immune response to cell stress. Cell 2006, 126, 659–662.
- Savio, L.E.B.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52.
- Bargiotas, P.; Krenz, A.; Hormuzdi, S.G.; Ridder, D.A.; Herb, A.; Barakat, W.; Penuela, S.; von Engelhardt, J.; Monyer, H.; Schwaninger, M. Pannexins in ischemia-induced neurodegeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 20772–20777.
- Wei, R.; Wang, J.; Xu, Y.; Yin, B.; He, F.; Du, Y.; Peng, G.; Luo, B. Probenecid protects against cerebral ischemia/reperfusion injury by inhibiting lysosomal and inflammatory damage in rats. Neuroscience 2015, 301, 168–177.
- Cisneros-Mejorado, A.; Gottlieb, M.; Cavaliere, F.; Magnus, T.; Koch-Nolte, F.; Scemes, E.; Perez-Samartin, A.; Matute, C. Blockade of P2X7 receptors or pannexin-1 channels similarly attenuates postischemic damage. J. Cereb. Blood Flow Metab. 2015, 35, 843–850.
- Freitas-Andrade, M.; Bechberger, J.F.; MacVicar, B.A.; Viau, V.; Naus, C.C. Pannexin1 knockout and blockade reduces ischemic stroke injury in female, but not in male mice. Oncotarget 2017, 8, 36973–36983.
- Prins, M.; Greco, T.; Alexander, D.; Giza, C.C. The pathophysiology of traumatic brain injury at a glance. Dis. Model Mech. 2013, 6, 1307–1315.
- Lumpkins, K.M.; Bochicchio, G.V.; Keledjian, K.; Simard, J.M.; McCunn, M.; Scalea, T. Glial fibrillary acidic protein is highly correlated with brain injury. J. Trauma 2008, 65, 778–782.
- Raghupathi, R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004, 14, 215–222.
- Meaney, D.F.; Smith, D.H. Cellular biomechanics of central nervous system injury. Handb. Clin. Neurol. 2015, 127, 105–114.
- Takano, T.; Oberheim, N.; Cotrina, M.L.; Nedergaard, M. Astrocytes and ischemic injury. Stroke 2009, 40, S8–S12.
- Blennow, K.; Brody, D.L.; Kochanek, P.M.; Levin, H.; McKee, A.; Ribbers, G.M.; Yaffe, K.; Zetterberg, H. Traumatic brain injuries. Nat. Rev. Dis. Primers 2016, 2, 16084.
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394.
- Weaver, L.C.; Bao, F.; Dekaban, G.A.; Hryciw, T.; Shultz, S.R.; Cain, D.P.; Brown, A. CD11d integrin blockade reduces the systemic inflammatory response syndrome after traumatic brain injury in rats. Exp. Neurol. 2015, 271, 409–422.
- Kenne, E.; Erlandsson, A.; Lindbom, L.; Hillered, L.; Clausen, F. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J. Neuroinflamm. 2012, 9, 17.
- Morganti, J.M.; Jopson, T.D.; Liu, S.; Riparip, L.K.; Guandique, C.K.; Gupta, N.; Ferguson, A.R.; Rosi, S. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J. Neurosci. 2015, 35, 748–760.
- Hsieh, C.L.; Niemi, E.C.; Wang, S.H.; Lee, C.C.; Bingham, D.; Zhang, J.; Cozen, M.L.; Charo, I.; Huang, E.J.; Liu, J.; et al. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J. Neurotrauma 2014, 31, 1677–1688.
- Junger, W.G. Immune cell regulation by autocrine purinergic signalling. Nat. Rev. Immunol. 2011, 11, 201–212.
- Alves, M.; Beamer, E.; Engel, T. The Metabotropic Purinergic P2Y Receptor Family as Novel Drug Target in Epilepsy. Front. Pharmacol. 2018, 9, 193.
- de Rivero Vaccari, J.P.; Bastien, D.; Yurcisin, G.; Pineau, I.; Dietrich, W.D.; De Koninck, Y.; Keane, R.W.; Lacroix, S. P2X4 receptors influence inflammasome activation after spinal cord injury. J. Neurosci. 2012, 32, 3058–3066.
- Ulmann, L.; Hatcher, J.P.; Hughes, J.P.; Chaumont, S.; Green, P.J.; Conquet, F.; Buell, G.N.; Reeve, A.J.; Chessell, I.P.; Rassendren, F. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J. Neurosci. 2008, 28, 11263–11268.
- Choo, A.M.; Miller, W.J.; Chen, Y.C.; Nibley, P.; Patel, T.P.; Goletiani, C.; Morrison, B., 3rd; Kutzing, M.K.; Firestein, B.L.; Sul, J.Y.; et al. Antagonism of purinergic signalling improves recovery from traumatic brain injury. Brain 2013, 136, 65–80.
- Garg, C.; Seo, J.H.; Ramachandran, J.; Loh, J.M.; Calderon, F.; Contreras, J.E. Trovafloxacin attenuates neuroinflammation and improves outcome after traumatic brain injury in mice. J. Neuroinflamm. 2018, 15, 42.
- Ni, B.K.; Cai, J.Y.; Lin, Q.; Zheng, K.H.; Lin, L.; Wu, J.H. Evaluation of serum pannexin-1 as a prognostic biomarker for traumatic brain injury. Clin. Chim. Acta 2019, 488, 159–164.


